 An official website of the United States government
An official website of the United States government
 ) or https:// means you've safely connected to the .mil website. Share sensitive information only on official, secure websites.
) or https:// means you've safely connected to the .mil website. Share sensitive information only on official, secure websites.
Tuberous Sclerosis Complex
A New Mouse Model Sheds Light on the Origin of Epilepsy in Tuberous Sclerosis Complex



Posted December 7, 2022
Xiaoping Wu, PhD; Alexander A. Sosunov, PhD; Wudu Lado, PhD; Jia Jie Teoh, PhD; Ahrom Ham, PhD; Hongyu Li, PhD; Osama Al Dalahmah, MD, PhD; Brian J.A. Gill, MD; Ottavio Arancio, MD, PhD; Catherine Schevon, MD, PhD; Wayne Frankel, PhD; Guy M. McKhann II, MD; David Sulzer, PhD; James E. Goldman, MD, PhD; Guomei Tang, PhD.
Columbia University
Tuberous Sclerosis Complex (TSC) is a genetic disorder caused by mutations in the TSC1 or TSC2 genes. This developmental disorder affects many systems of the body and is usually characterized by epilepsy, autism, and cognitive impairments, including long-term and working memory deficits. A key, unresolved issue is the cause of the neurological symptoms in TSC patients. A group of investigators at Columbia University have recently published an article in Cell Reports on a new mouse model of TSC that sheds light on the cause of epilepsy in TSC patients. Among them, Drs. Tang, Sulzer, and Goldman have received Idea Development Awards (IDA) in differing years from the TSC Research Program (TSCRP) of the Congressionally Directed Medical Research Programs under the Department of Defense (DOD).
 From left to right, Dr. Guomei Tang, Dr. James E. Goldman, Dr. David Sulzer,
From left to right, Dr. Guomei Tang, Dr. James E. Goldman, Dr. David Sulzer, Dr. Wudu Lado, Columbia University (Photos provided)
Dr. Sulzer was awarded a Fiscal Year 2011 (FY11) TSCRP IDA to investigate altered astrocyte-neuron interactions in TSC and a potential role for these pathological changes in the epileptogenesis observed in this disorder. This award supported the study of astrocytic mechanisms in pruning excessive excitatory synapse during development which may produce neuronal hyperexcitability.
Dr. Goldman's FY14 IDA further studied the molecular mechanisms underlying epileptogenesis and seizure progression in Tsc1 deficient mouse models. Using systems biology/gene expression profiling and electrophysiological, biochemical, immunohistochemical, and behavioral studies, Dr. Goldman's group focused on the molecular signatures that are significantly changed during epileptogenesis in the model mice.
Dr. Tang's FY15 IDA delved into the altered mTOR-related macroautophagy and its role in TSC-associated neurocognitive deficits (mTOR is a kinase that is activated in TSC and in turn suppresses autophagy, a process by which cells can degrade their own components). The team also worked together to shed light on how epilepsy might originate in TSC.
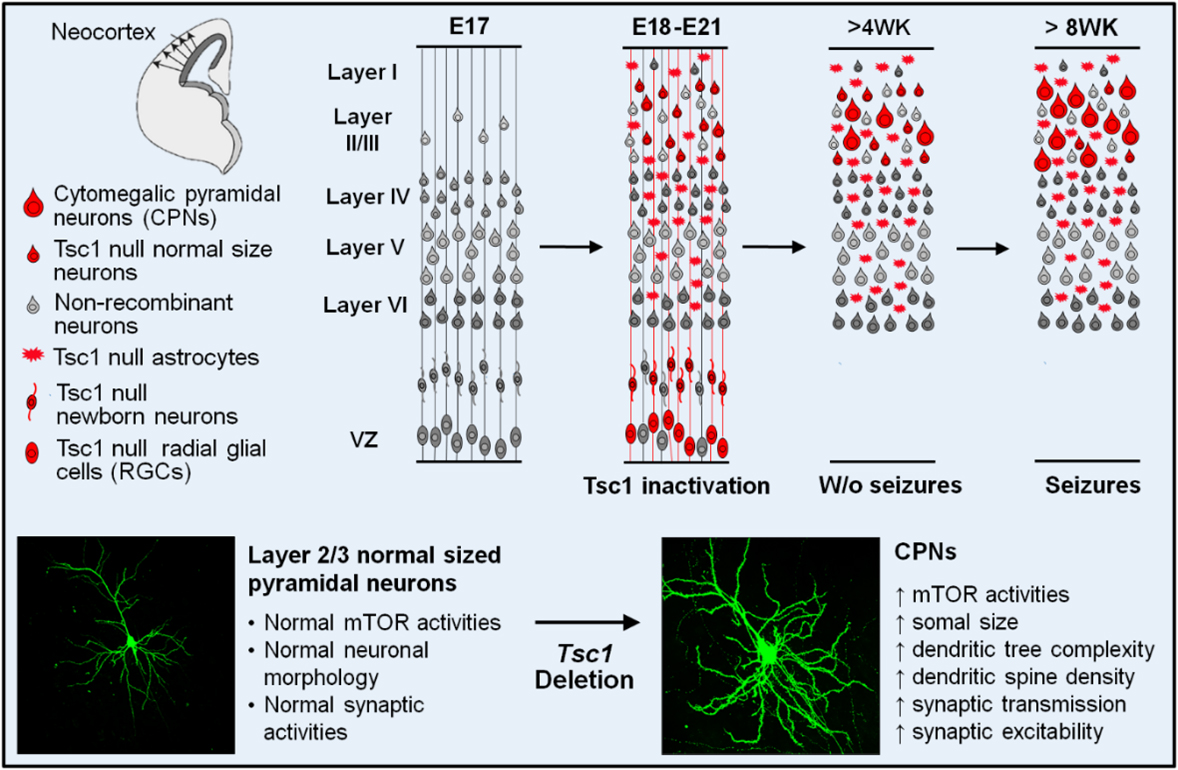
Figure 1: The team developed a new Tsc1 conditional knockout (Tsc1CKO) mouse model. In this model,
The recent multi-laboratory collaborative publication titled, "Synaptic hyperexcitability of cytomegalic pyramidal neurons contributes to epileptogenesis in tuberous sclerosis complex," provides evidence that the new mouse model, developed by this team, is characterized by greatly enlarged cerebral cortical neurons and those cells are epileptogenic. The mice exhibited social and cognitive impairment and spontaneous seizures. The team utilized a conditional knock-out system in which the Tsc1 gene is inactivated only in specific embryonic cell types. Tsc1 depletion occurs in a subset of cortical neurons, leading to the development of enlarged ("cytomegalic") pyramidal neurons that mimic the neurons having abnormal morphological characteristics seen in human patients with TSC. These neurons show aberrant overgrowth of their cellular processes, enhanced excitatory synaptic transmission, and increased susceptibility to seizure-like activities. Heightened synaptic excitation contributes to the observed cortical hyperexcitability and epileptogenesis.
To follow up this study, the team received an FY20 Exploration - Hypothesis Development Award (EHDA) award (Principal Investigator: Dr. Lado) to investigate whether the dysfunction of a neuronal specific chloride transporter enhances the synaptic excitability of cytomegalic neurons in TSC. The team is also working on a follow-up article comparing the pathological characteristics of the enlarged mouse neurons to the dysplastic neurons found in human tubers, with the goal to identify common cellular and molecular changes that are involved in epileptogenesis.
"Our studies have given us new insights into the pathological and clinical features of TSC and opened up future studies to go deeper into molecular mechanisms," Dr. Goldman said. "We are all grateful for the DOD support of our efforts."
Links:
Last updated Friday, December 12, 2025