
Tuberous Sclerosis Complex
Building Protein Network Models of Epilepsy to Find New Treatments in TSC
Posted September 22, 2017
Kimberly Raab-Graham, Ph.D., University of Texas at Austin*

University of Texas at Austin*
The neurological symptoms associated with tuberous sclerosis complex (TSC), such as epilepsy, autism spectrum disorder (ASD), and intellectual disability, are those reported to have the greatest impact on the lives of patients and caregivers. While still a point of debate, there is growing evidence that infantile seizures may contribute to the developmental delays and/or ASD in TSC patients, suggesting that stopping the seizures may be able to prevent the cognitive problems that develop at a later time point. Nearly 80-90% of infants with TSC have epilepsy. There is no cure for TSC, and treatments are limited to drugs targeting the prevalent seizures. The lack of effective treatments means that TSC patients face a lifelong risk of debilitating seizures and neurological consequences.
With funding from a Tuberous Sclerosis Complex Research Program (TSCRP) Idea Development Award, Dr. Kimberly Raab-Graham and her group are providing new insight into the development of TSC. TSC has historically been considered a disorder where the brain makes too much protein. Dr. Raab-Graham's research has shown that brain cells (neurons) may not make the "right" proteins, and some of the proteins her team has identified may be protective against epilepsy.
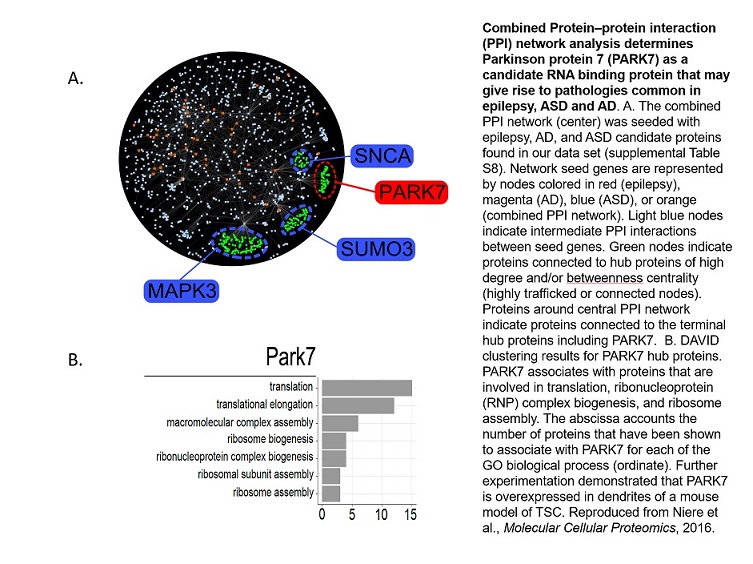
Dr. Raab-Graham's lab investigated how and why effective therapies for TSC-related epilepsy work, in order to gain a better understanding of the disease mechanism. In this case, they used rapamycin as a tool to ask the simple question, "How does the synapse change (i.e., the site of communication between two neurons) when mTOR is turned off?" Using a combination of mass spectrometry and bioinformatics, Dr. Raab-Graham discovered that 75% of the proteins at the synapse change with rapamycin. Using these data as a starting point to build a network of interacting proteins (much like how tinker toys are assembled), the group identified an RNA binding protein, Parkinson's 7, as a protein that regulates the expression of proteins that are linked to epilepsy.
Finding medications to prevent seizures in TSC before they start is critical. To that end, Dr. Raab-Graham's group plans to identify all of the "epileptic" proteins that are regulated by Parkinson's 7 and determine how their expression is disrupted in a mouse model of TSC. In addition, Dr. Raab-Graham shares, "We realize it is vital to develop preclinical models of TSC that allow us to rapidly screen for the contribution of these "epileptic" proteins to seizure generation and to actively seek repurposed or new medications to prevent seizures." To accomplish this, Dr. Raab-Graham is collaborating with Dr. Dwayne Godwin's lab at Wake Forest School of Medicine and the Wake Forest Baptist Health's Comprehensive Epilepsy Center.
"We are really excited about the broad implications that our research may have," Dr. Raab-Graham states. "While TSC is considered a rare disease, common symptoms shared with other neurological disorders, for example seizures, have allowed us to identify new pharmacological targets for TSC. The implications of our findings may impact other neurological disorders even as seemingly different as Alzheimer's Disease and traumatic brain injury."
Link:
* Currently at Wake Forest University School of Medicine
Last updated Wednesday, March 12, 2025