
Posted November 12, 2014
Brendan Manning, Ph.D., Harvard University
 Tuberous sclerosis complex (TSC) is a rare genetic disease that causes the formation of widespread benign tumors predominantly in the brain, skin, kidneys, heart, and lung. Genetically, TSC is caused by functional loss of either the TSC1 or TSC2 gene leading to elevated activation of the mTORC1 gene complex signaling pathway, which causes synthesis of proteins that lead to the aberrant cell growth of TSC tumors. Use of the mTORC1 inhibitor rapamycin has shown promising results for the treatment of TSC tumors, although the tumors often regrow after treatment has ceased, presenting a need for the development of new, improved therapies. To accomplish this, Dr. Brendan Manning at Harvard University sought to further characterize the regulation and function of mTORC1 in the context of TSC1/2 gene loss.
Tuberous sclerosis complex (TSC) is a rare genetic disease that causes the formation of widespread benign tumors predominantly in the brain, skin, kidneys, heart, and lung. Genetically, TSC is caused by functional loss of either the TSC1 or TSC2 gene leading to elevated activation of the mTORC1 gene complex signaling pathway, which causes synthesis of proteins that lead to the aberrant cell growth of TSC tumors. Use of the mTORC1 inhibitor rapamycin has shown promising results for the treatment of TSC tumors, although the tumors often regrow after treatment has ceased, presenting a need for the development of new, improved therapies. To accomplish this, Dr. Brendan Manning at Harvard University sought to further characterize the regulation and function of mTORC1 in the context of TSC1/2 gene loss.
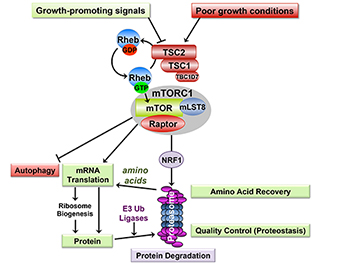
With support from a Fiscal Year 2009 Tuberous Sclerosis Complex Research Program Idea Development Award, Dr. Manning utilized cells lacking either the TSC1 or TSC2 gene and found that these cells had a 20%-25% increase in protein synthesis that was dependent on mTORC1. Surprisingly, he also found that these cells had an mTORC1-dependent increase in protein degradation, which does not occur in cells with intact TSC1 or TSC2 genes. Dr. Manning hypothesizes that this novel mechanism of increased protein degradation in cells with high rates of protein synthesis due to mTORC1 activation serve two purposes: (1) as a quality control mechanism to prevent the accumulation of mis-folded proteins and (2) to assure that such cells do not starve themselves of the amino acid building blocks required to make new proteins.
Upon further investigation into this novel discovery, Dr. Manning found that loss of the TSC2 gene triggered a signaling cascade that activated mTORC1 and stimulated expression of every component of the proteasome, a large cellular complex that is responsible for the degradation of unneeded or damaged proteins. His laboratory determined that a gene called NRF1 is induced by mTORC1 activation and is required for the increased expression of proteasome genes in TSC2-deficient cells or upon stimulation of normal cells with growth factors. Their study further confirmed that this mTORC1-mediated activation of NRF1 and the proteasome occurs in the enlarged neurons of a TSC mouse model involving a neuron-specific mutation of TSC2. Importantly, he found that TSC2-deficient cells were especially sensitive to NRF1 inhibition, demonstrating that this response accompanying mTORC1's established role in promoting protein synthesis is required for survival of TSC2 mutant cells. This novel vulnerability makes NRF1 a potential new therapeutic target for TSC.
Taken together, these results provide new insight into the molecular mechanisms involved with TSC1/2 gene functional loss that ultimately leads to TSC tumors. Further understanding of these mechanisms provides the basis for identifying novel therapeutic targets, including NRF1, and the development of new therapies for individuals with TSC.
Publications:
Zhang Y, Nicholatos J, Dreier J, et al. 2014. Coordinated regulation of protein synthesis and degradation by mTORC1. Nature 513(7518):440-443.
Dibble CC and Manning BD. 2013. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nature Cell Biology 15:555-564.
Ricoult SJ and Manning BD. 2013. The multifaceted role of mTORC1 in the control of lipid metabolism. European Molecular Biology Organization Report 14(3):242-251.
Auricchio N, Malinowska I, Shaw R, et al. 2012. Therapeutic trial of metformin and bortezomib in a mouse model of tuberous sclerosis complex (TSC). PLoS One 7(2):e31900.
Links: