- Newly Discovered Tumor Suppressor Gene May Lead to Earlier Diagnosis of Aggressive Prostate Cancer
- The Power of Antioxidants: A Vitamin E Derivative Targets Androgen-Induced Oxidative Stress and Prostate Cancer Growth
- Critical Role of the PCRP in FDA Approval of Abiraterone Acetate
- SPINK1: A Promising New Target in Prostate Cancer
- A Monoclonal Antibody Targeting N-Cadherin Inhibits Prostate Cancer Growth, Metastasis, and Castration Resistance
- Prostate Cancer Skeletal Metastases: Insight into Pathobiology and Interventions
- Predoctoral Training Award Recipient Develops New Method to Study Prostate Cancer Cell Survival
Newly Discovered Tumor Suppressor Gene May Lead to Earlier Diagnosis of Aggressive Prostate Cancer
Posted December 20, 2011
Lloyd Trotman, Ph.D., Cold Spring Harbor Laboratory, Cold Spring Harbor, New York
Since most newly diagnosed prostate cancers are not considered lethal, a high priority for the field of prostate cancer research is to create predictive tests that can identify men who, at the time of diagnosis, are at high risk for developing aggressive disease. Recent advances in understanding the human prostate cancer genome, along with the development of more informative animal models, are providing key clues to the genetic changes that may be able to distinguish indolent from aggressive prostate cancer. Dr. Lloyd Trotman, using support from a Fiscal Year 2008 Prostate Cancer Research Program New Investigator Award, has identified a new tumor suppressor gene, PHLPP1 (pronounced "flip one"), which cooperates with another well-known prostate cancer tumor suppressor gene, PTEN, to prevent progression to aggressive prostate cancer. The PTEN protein, as a phosphatase, works by removing phosphate molecules from its targets and stops cell proliferation by preventing activation of the PI3 Kinase/AKT pathway. PHLPP1 is also a phosphatase recently identified as a deactivator of AKT and also functions as a suppressor of prostate tumors.
Importantly, Dr. Trotman discovered that the PHLPP1 gene was deleted in about 15% of patients with tumors localized to the prostate gland and in about 40% of patients whose tumors had metastasized to other parts of the body. Moreover, most of these metastatic tumors had also lost PTEN. Although PTEN was deleted in localized disease to a similar degree as PHLPP1, local tumors from individual patients only had deletions of one or the other gene, seeming to imply that the deletion of both genes may lead to the development of aggressive disease. Regarding prognosis, he found that low RNA levels of PHLPP1 and PTEN corresponded with shorter relapse-free survival times after radical prostatectomy.
So, to better understand the role of PHLPP1 and PTEN deletions in the progression of prostate cancer to metastatic disease, Dr. Trotman genetically engineered a mouse model lacking the PHLPP1 gene. He found that loss of PHLPP1 alone caused a pre-malignant form of prostate cancer (high-grade prostatic intraepithelial neoplasia) in the mice, similar to mice lacking the PTEN gene. Mice lacking both genes, however, rapidly developed fast-growing prostate cancer similar to patients with the PHLPP1 and PTEN co-deletion. Since both genes act as brakes on cell growth mediated by the Akt signaling pathway, Dr. Trotman analyzed Akt activity in the mouse prostate and found an increase in Akt activity and cell growth at 4 months of age, followed by a dramatic burst of both activities at 8-12 months of age, precisely when prostatic lesions were forming. To determine the cause of the sudden burst of Akt activity, Dr. Trotman conducted additional experiments and found that a third tumor suppressor gene, p53, had lost functionality, which coincided with cancer progression. These data suggest that the loss of these three tumor suppressor genes triggers a time-dependent path from slow-growing prostate cancer to fast-growing, aggressive disease. Importantly, he found that every PTEN and PHLPP1 co-deleted human metastasis sample also had p53 deletion, thus validating the new concept.
Taken together, these results for the first time identify a critical role for PHLPP1 in prostate cancer. Furthermore, the ability to define the status of PHLPP1 and PTEN in prostate cancer biopsies, especially at the RNA level, could enable clinicians to identify patients who are at risk of developing aggressive disease and who might benefit from therapies targeting the Akt pathway, several of which are currently being tested in clinical trials.Haploinsufficiency and senescence in prostate cancer progression
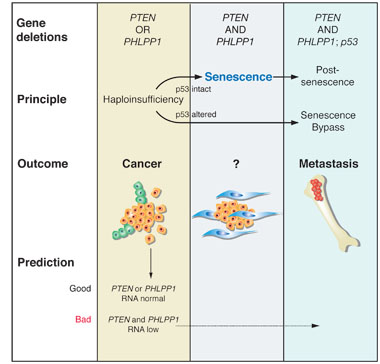
1. Genetics of progression: Co-deletion of PTEN and PHLPP1 is restricted to metastasis and associated with p53 deletion.
2. Progression principle: Haploinsufficiency1 for PTEN or PHLPP1 results in cancer. Combined loss and complete loss of PTEN can trigger senescence arrest2 if p53 is intact to function as a barrier against progression.
3. Outcomes associated with the above principles or alterations are indicated.
4. Predictive value of the principle: Low RNA levels for PTEN and PHLPP1 can be found in some surgical samples and are associated with poor prognosis.
1Haploinsufficiency refers to the condition of a cell containing only a single functional copy of a gene that normally has two functional copies.
2Senescence arrest is a mechanism through which cells encounter a barrier to proliferation and stop dividing.Publications:
Chen M, Pratt CP, Zeeman ME, Schultz N, Taylor BS, O'Neill A, Castillo-Martin M, Nowak DG, Naguib A, Grace DM, Murn J, Navin N, Atwal GS, Sander C, Gerald WL, Cordon-Cardo C, Newton AC, Carver BS, Trotman LC. 2011. Identification of PHLPP1 as a Tumor Suppressor Reveals the Role of Feedback Activation in PTEN-Mutant Prostate Cancer Progression. Cancer Cell 20(2):173-186.
Links:
The Power of Antioxidants: A Vitamin E Derivative Targets Androgen-Induced Oxidative Stress and Prostate Cancer Growth
Posted July 29, 2011
George Wilding, M.D., University of Wisconsin, Madison, Wisconsin
Sometimes things are worth waiting for, and this is turning out to be quite true for the innovative, basic research ideas supported by the Prostate Cancer Research Program (PCRP), even in its earliest years of existence. In fiscal year 1997, the first year that the PCRP offered funding to the research community, Dr. George Wilding of the University of Wisconsin won a PCRP Idea Development Award with his idea to develop an understanding of how antioxidants might be useful as chemopreventative agents for prostate cancer. He proposed to investigate the molecular mechanisms by which androgens increase reactive oxygen species (ROS). This early research has now led to the development of a novel new drug, APC-100, a derivative of vitamin E, which is entering clinical trials for the treatment of prostate cancer.
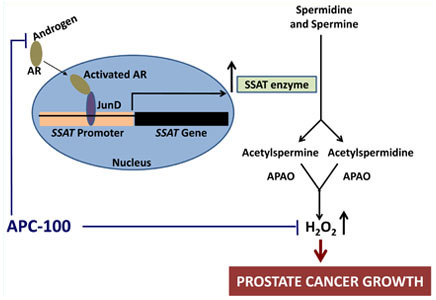
ROS are chemically-reactive molecules, such as hydrogen peroxide (H2O2), that contain and release oxygen, which can cause damage to the cellular structures of DNA, proteins, and membranes when it is overproduced in cells. ROS are believed to a play key role in both initiation and progression of prostate cancer. Dr. Wilding showed that, in the presence of androgen, the androgen receptor in prostate cancer cells binds to the transcription factor JunD and activates the expression of the spermidine/spermine N1-acetyl transferase gene (SSAT). SSAT controls the first step in the breakdown of polyamines (spermidine and spermine), resulting in H2O2 as a by-product of this metabolic process. A hallmark of the prostate gland is the large amount of polyamines produced by prostatic epithelial cells in the generation of prostatic fluid. Such high levels of polyamines are not seen in other human tissues. Consequently, the induction of polyamine oxidation by the SSAT pathway results in very high levels of ROS and ROS-mediated damage in prostate cancer cells. This observation implicates the SSAT-polyamine oxidation pathway as a major mechanism through which androgen induces ROS in prostate cancer cells.
Many plant oils such as soy, corn, and olive oil contain vitamin E, which is thought to offer anti-oxidant protection against ROS. Dr. Wilding therefore tested the anti-oxidant effects of vitamin E in prostate cancer cells by treating them with vitamin E derivatives. He found that, rather than acting directly against ROS, the anti-oxidant moiety of vitamin E (now called APC-100) interacts directly with the androgen receptor, abrogates androgen-inducing gene expression, and blocks cellular damage caused by ROS. The upshot is that APC-100 inhibits both the growth and viability of both androgen-dependent and -independent prostate cancer cells. Pre-clinical studies in animal models of androgen-dependent and -independent prostate cancer confirmed these results and demonstrated that APC-100 delays tumor progression and increases survival. In June 2011, the U.S. Food and Drug Administration accepted an Investigational New Drug application for APC-100, which will soon enter a Phase I/II clinical trial in men with castrate-resistant prostate cancer at the University of Wisconsin Carbone Cancer Center.
Schematic diagram showing a possible mechanism of androgen-induced increase in cellular ROS production in prostate cancer cells through the AR-JunD complex and inhibition of this pathway by anti-androgenic anti-oxidant APC-100. AR: androgen receptor; APAO: N'-acetyl polyamine oxidase; H2O2: hydrogen peroxide. Adapted from Mehraein-Ghomi F, Basu HS, Church DR, Hoffmann FM, Wilding G. 2010. Androgen receptor requires JunD as a coactivator to switch on an oxidative stress generation pathway in prostate cancer cells. Cancer Research 70(11):4560-4568.
Publications:
Mehraein-Ghomi F, Basu HS, Church DR, Hoffmann FM, Wilding G. 2010. Androgen receptor requires JunD as a coactivator to switch on an oxidative stress generation pathway in prostate cancer cells. Cancer Research 70(11):4560-4568.
Basu HS, Thompson TA, Church DR, Clower CC, Mehraein-Ghomi F, Amlong CA, Martin CT, Woster PM, Lindstrom MJ, Wilding G. 2009. A small molecule polyamine oxidase inhibitor blocks androgen-induced oxidative stress and delays prostate cancer progression in the transgenic adenocarcinoma of the mouse prostate model. Cancer Research 69(19):7689-7695.
Thompson TA and Wilding G. 2003. Androgen antagonist activity by the antioxidant moiety of vitamin E, 2,2,5,7,8-pentamethyl-6-chromanol in human prostate carcinoma cells. Molecular Cancer Therapeutics 2(8):797-803.
Links:
Critical Role of the PCRP in FDA Approval of Abiraterone Acetate
Posted July 14, 2011
Prostate cancer patients and physicians have had good reason for increased hope this year. Along with last year's US Food and Drug Administration (FDA) approval of the new prostate cancer drugs PROVENGE, JEVTANA, and XGEVA, in April 2011 the FDA also approved the use of abiraterone acetate (marketed as ZYTIGA) for patients with the most lethal form of prostate cancer: metastatic, castration-resistant prostate cancer (mCRPC). Abiraterone acetate is a potent inhibitor of a specific enzyme, CYP17A1, found in the testes, adrenal gland, and prostate tumors, that is crucial for the synthesis of testosterone (an androgen), which drives the growth of prostate cancer. The Prostate Cancer Clinical Trials Consortium (PCCTC), made possible through joint funding support from the Prostate Cancer Research Program (PCRP) and the Prostate Cancer Foundation (PCF), was critical to completing the phase I and II clinical trials that helped to accelerate abiraterone acetate to approval in record time and provide a new standard treatment for prostate cancer patients.
Abiraterone first entered the PCCTC in 2006 by lead investigator Dr. Charles Ryan of the University of California, San Francisco, in a phase I clinical trial in patients with mCRPC who had previously received chemotherapy. This trial established that the dosage of abiraterone was tolerated by patients at all levels tested and demonstrated promising outcomes, such as decreased androgen levels, suggesting activity against mCRPC. Soon afterward, Dr. Ryan and PCCTC director Dr. Howard Scher of Memorial Sloan-Kettering Cancer Center led two phase II clinical trials in mCRPC patients who had failed androgen deprivation therapy and chemotherapy. These trials, completed in 2008, further demonstrated antitumor effects such as declines in prostate specific antigen (PSA) levels in patients and delays in time to biochemical progression (as judged by increasing PSA levels).
These successful phase I and II clinical trials led directly to the international phase III clinical trial upon which the FDA based its approval of abiraterone acetate. This trial, guided by Dr. Scher and Dr. Johann de Bono of the Royal Marsden Hospital in London, started in mid-2008 and studied 1,195 patients with mCRPC who had previously been treated with prednisone. The results confirmed an increased patient survival in the abiraterone acetate group compared to placebo (14.8 months versus 10.9 months, respectively), PSA declines (38% versus 10.1%, respectively), increased time to biochemical progression (10.2 months versus 6.6 months, respectively), and side-effects that were easily tolerated by patients. The PCCTC's successful acceleration of abiraterone to FDA approval in record time is another demonstration of the Consortium's ability to work together as a team to streamline the medical, ethical, scientific, institutional, legal, and regulatory complexities of the clinical trial process for the benefit of prostate cancer patients. Additional phase II clinical investigations by the PCCTC are ongoing to improve abiraterone's effectiveness and determine whether it will benefit men with diverse treatment histories and stages of prostate cancer.
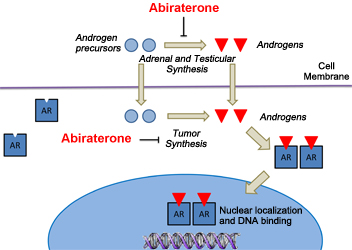
Abiraterone inhibits androgen synthesis in prostate cancer cells. Androgen precursors and androgens enter the prostate cancer cell, where the precursors are converted to androgens ("Tumor Synthesis"). The newly synthesized androgens, as well as those made in the adrenal glands and testes, bind to the androgen receptor and are transported to the nucleus, where they bind to DNA and turn on genes controlling prostate cancer cell growth. Abiraterone blocks androgen synthesis at all three sites.
Publications:
Ryan CJ, Shah SK, Efstathiou E, Smith MR, Taplin ME, Bubley GJ, Logothetis CJ, Kheoh T, Kilian C, Haqq C, Molina A, Small EJ. 2011. Phase II study of abiraterone acetate in chemotherapy-naive metastatic castration-resistant prostate cancer displaying bone flare discordant with serologic response. Clinical Cancer Research [June; Epub ahead of print]
Danila DC, Morris MJ, de Bono JS, Ryan CJ, Denmeade SR, Smith MR, Taplin M-E, Bubley GJ, Kheoh T, Haqq C, Molina A, Anand A, Koscuiszka M, Larson SM, Schwartz LH, Fleisher M, and Scher HI. 2010. Phase II multicenter study of abiraterone acetate plus prednisone therapy in patients with docetaxel-treated castration-resistant prostate cancer. Journal of Clinical Oncology 28:1496-1501.
Ryan CJ, Smith MR, Fong L, Rosenberg JE, Kantoff P, Raynaud F, Martins V, Lee G, Kheoh T, Kim J, Molina A, and Small EJ. 2010. Phase I clinical trial of the CYP17 inhibitor abiraterone acetate demonstrating clinical activity in patients with castration-resistant prostate cancer who received prior ketoconazole therapy. Journal of Clinical Oncology 28:1481-1488.
Links:
SPINK1: A Promising New Target in Prostate Cancer
Posted June 16, 2011
Arul Chinnaiyan, M.D., Ph.D., University of Michigan, Ann Arbor, Michigan
In 2005, Dr. Arul Chinnaiyan and his team published a landmark discovery in prostate cancer. They had discovered that over 50% of prostate cancer patients harbor gene fusions (TMPRSS2-ETS) in their tumors, and that these gene fusions likely cause prostate cancer. This discovery was a major paradigm shift, as gene fusions had never before been discovered in any solid tumor, let alone prostate tumors. Building on his seminal discovery, Dr. Chinnaiyan decided to investigate other genetic changes that could be critical to prostate cancer development, and that consequently might pose prime targets for the development of new and more effective prostate cancer therapies.
With a Fiscal Year 2007 Idea Development Award from the Prostate Cancer Research Program (PCRP), Dr. Chinnaiyan's team used computation-based analyses of prostate cancer gene expression datasets to identify a gene called SPINK1 (serine protease inhibitor, Kazal type 1), which they found to be overexpressed in 10% of prostate cancers and, moreover, that this group of SPINK1-positive tumors were found in patients with more aggressive tumors than patients with SPINK1-negative tumors. Thus, SPINK1 expression may represent a novel molecular subtype of prostate cancer that is more aggressive than SPINK1-negative cancers and may respond best to distinct treatment regimens.
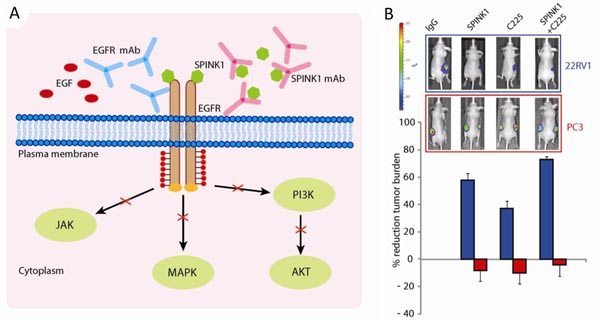
To understand how SPINK1 contributes to prostate cancer, the investigators treated normal and cancerous prostate cells with SPINK1 protein and observed increased proliferation and invasiveness of both cell types. They also showed that SPINK1 activates the epidermal growth factor receptor (EGFR) on the surface of prostate cancer cells, leading to cell growth. Using a mouse model of SPINK1-positive prostate cancer (22RV1 cells), they showed that monoclonal antibodies to either SPINK1 or EGFR (cetuximab) could slow the growth of SPINK1-positive tumors by over 60% and 40%, respectively. When used in combination, the antibodies slow the growth of SPINK1-positive tumors by 75% while having no effect on SPINK1-negative tumors. These results, while quite promising, will also require testing in the background of other genetic alterations, which may influence the response to therapeutic treatment.
This work highlights the dedication of PCRP investigators in addressing the major challenges regarding prostate cancer, which include (1) distinguishing aggressive from indolent disease and (2) developing effective treatments for advanced prostate cancer. A new era of personalized medicine for prostate cancer patients appears to be on the horizon as new biomarkers are identified to subclassify tumor types, better guide treatment decision-making, and provide optimal outcomes for prostate cancer patients.
A) Schematic illustration showing how Spink1 and EGFR monoclonal-antibodies block EGFR to inhibit SPINK1-positive prostate cancer cell growth.
B) Bioluminescence images of mice with SPINK1-positive 22RV1 tumors (blue) or SPINK1-negative PC3 tumors (red) treated with control (IgG), SPINK1, and/or EGFR (C225) monoclonal-antibodies; the bar graph below shows the percent reduction in tumor burden for each treatment.Publication:
Ateeq B, Tomlins SA, Laxman B, Asangani IA, Cao Q, Cao X, Li Y, Wang X, Feng FY, Pienta KJ, Varambally S, Chinnaiyan AM. 2011. Therapeutic targeting of SPINK1-positive prostate cancer. Science Translational Medicine 3(72):72ra17.
Links:
Public and Technical Abstracts: Characterization of SPINK1 in Prostate Cancer
A Monoclonal Antibody Targeting N-Cadherin Inhibits Prostate Cancer Growth, Metastasis, and Castration Resistance
Posted May 10, 2011
Robert Reiter, M.D., and Matthew Rettig, M.D., University of California, Los Angeles
With support from a Fiscal Year 2008 Synergistic Idea Development Award through the Department of Defense Prostate Cancer Research Program, Drs. Robert Reiter and Matthew Rettig recently published an article in Nature Medicine describing their study of an antibody that may offer substantial therapeutic improvement and slow the progression of prostate cancer. This antibody targets a protein, N-cadherin, on the surface of metastatic prostate cancer cells.
Radical prostatectomy and radiation therapy are effective treatments for primary prostate cancer, although some patients will experience disease recurrence. Standard treatment for recurrent prostate cancer is androgen deprivation therapy (ADT), which is initially very effective in causing the tumors to shrink and disappear. Over time (months to years), however, tumors become resistant to this therapy and re-emerge in an androgen-independent form (i.e., capable of growing in the apparent absence of androgens) called castration-resistant prostate cancer (CRPC). These tumors will metastasize to other tissues, particularly bone.
Molecular events underlying the transition from androgen-dependent to castration-resistant prostate cancer are poorly understood. To identify pathways that contribute to castration resistance, Drs. Reiter and Rettig compared gene expression patterns of androgen-dependent prostate cancer cells transplanted in either castrated or matched androgen-dependent xenograft mice. The tumors that developed from the transplanted cells in castrated mice were removed and sequentially transplanted into other castrated mice. The investigators found that the tumors became increasingly metastatic and the expression of one gene, a cell surface protein called N-cadherin, rose consistently with each subsequent passage. Moreover, high levels of N-cadherin expression were found in tumor tissues of patients treated with ADT and patients with CRPC, compared with very low levels of N-cadherin in patients with androgen-dependent prostate cancer. These data suggest that increased N-cadherin expression is involved in the development of castration resistance in prostate cancer patients.
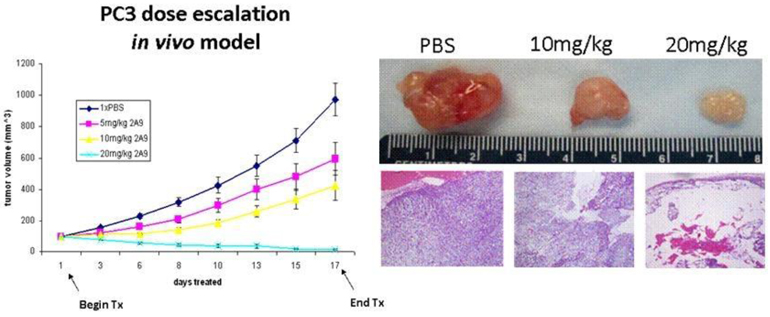
To test whether N-cadherin could be a therapeutic target in CRPC, monoclonal antibodies against N-cadherin were developed and used to treat mice with CRPC tumors. Drs. Reiter and Rettig found that the N-cadherin antibody decreased tumor growth by an average of 70%. Additionally, N-cadherin antibody treatment prevented tumor invasion and metastasis whereas untreated mice experienced lymph node metastasis. Importantly, mice with established androgen-dependent prostate tumors that were treated with both castration and N-cadherin antibody showed a dramatic delay in time to progression to CRPC compared to mice treated with castration (ADT) alone. These results show that N-cadherin is important for the development of metastatic CRPC and that therapeutic targeting of this protein may have significant clinical benefit.
Treatment with higher doses of 2A9 N-cadherin antibody results in tumor regression. Mice were injected with PC3 cells subcutaneously and when the tumors measured approximately 100 mm3 in volume, the mice were treated with PBS (control), 5, 10, or 20 mg/kg of 2A9 antibody. Mice treated with 20 mg/kg of the antibody showed significant tumor regression, which is visualized graphically (left). The excised tumors are shown (upper right), and H&E staining of the excised tumors show considerable hypocellularity (lower right).
Publication:
Tanaka H, Kono E, Tran CP, Miyazaki H, Yamashiro J, Shimomura T, Fazli L, Wada R, Huang J, Vessella RL, An J, Horvath S, Gleave M, Rettig MB, Wainberg ZA, and Reiter RE. 2010. Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nature Medicine 16(12):1414-1420.
Links:
Prostate Cancer Skeletal Metastases: Insight into Pathobiology and Interventions
Posted February 17, 2011
Evan Keller, D.V.M., Ph.D., University of Michigan, Ann Arbor, Michigan
Recently, the U.S. Food and Drug Administration (FDA) approved denosumab for treatment of bone loss during prostate cancer treatment based, in part, on preclinical studies by Dr. Evan Keller, recipient of two separate grants awarded by the Department of Defense Prostate Cancer Research Program (PCRP). Through his work, Dr. Keller has demonstrated that by inhibiting a key regulator of bone resorption, RANKL (receptor activator of NF-kB ligand), one could slow the progression of prostate cancer bone metastases.
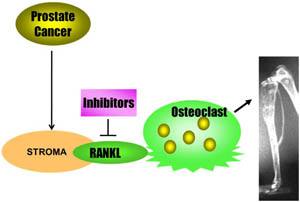
Bone metastases are common in prostate cancer, occurring in up to 80% of patients with advanced disease and causing severe skeletal complications (i.e. pain, fractures, and spinal cord compression), which contribute to prostate cancer morbidity and mortality. In addition, bone integrity is compromised as a result of androgen deprivation therapy (ADT), a common treatment for advanced prostate cancer. ADT decreases bone mineral density (BMD), leading to increased risk of bone fractures. Bone is a dynamic tissue that is constantly being remodeled by the opposing actions of resorption and formation, performed by cells in the bone known as osteoclasts and osteoblasts, respectively. This process is normally controlled by three key regulatory proteins: (1) RANKL, which is secreted by bone marrow stromal cells and osteoblasts; (2) RANK (receptor activator of NF-kB), which is located on the surface on osteclasts that binds RANKL and induces bone resorption; and (3) OPG (osteoprotegerin), which is secreted by osteoblasts and functions as a "decoy receptor" by competing with RANK for RANKL binding, thus preventing osteoclast-induced bone resorption.
In experiments funded by his PCRP awards, Dr. Keller demonstrated through the use of orthotopic models of prostate cancer bone metastases (i.e. experimentally established bone tumors) that prostate cancer cells stimulate RANKL secretion in the bone microenvironment, causing osteclastic bone resorption. He showed that treating mice with a synthetic fusion protein (i.e., containing amino-terminus of OPG fused to the Fc portion of the immunoglobulin heavy chain [OPG-Fc]) could inhibit the establishment of prostate cancer in bone. Upon finding that OPG can block apoptosis of prostate cancer cells, Dr. Keller reasoned that OPG-Fc might not be clinically useful; therefore, he subsequently tested the effects of another RANKL inhibitor. The evaluation of soluble RANK-Fc (sRANK-Fc, obtained from Amgen) showed that it reduced prostate cancer-induced bone lesions in mice as assessed by several means including radiography, bone marrow density measurements, and histopathology.
Dr. Keller's preclinical results demonstrated that inhibition of RANKL diminishes the establishment and progression of prostate cancer in bone, which suggests that inhibiting RANKL in men with skeletal metastases will slow their tumor progression. As a result of Dr. Keller's research, Amgen has since developed a human monoclonal antibody directed against RANKL. In November 2010, the FDA approved denosumab (XGEVATM ) for treatment of bone loss during cancer treatment; this approval was based on three clinical trials that demonstrated the drug's effectiveness over the current standard of care.
Prostate cancer cells alter the stromal cell population within the bone to produce RANKL. The increased production of RANKL stimulates osteoclast production and activity, promoting the osteolytic component of prostate cancer bone metastases.
Publications:
Zhang J, Dai J Smith P, Qi Y, Lin D, Strayhorn C, Mizokami A, Fu Z, and Keller ET. 2001. Osteoprotegerin inhibits prostate cancer-induced osteoclastogenesis and prevents prostate tumor growth in the bone of mice. J Clin Invest 107:1235-1244.
Zhang J, Dai J, Yao Z, Lu Y, Dougall W, Keller ET. 2003. Soluble receptor activator of nuclear factor kappaB Fc diminishes prostate cancer progression in bone. Cancer Res 63(22):7883-90.
Links:
Public and Technical Abstracts: Prostate Cancer Skeletal Metastases: Pathobiology and Interventions
Predoctoral Training Award Recipient Develops New Method to Study Prostate Cancer Cell Survival
Posted January 27, 2011
Laura Lamb, Ph.D., Van Andel Research Institute, Grand Rapids, Michigan
Since growth of prostate cancer cells is primarily driven by androgens interacting with androgen receptor (AR) to activate signaling pathways for cell proliferation, androgen ablation therapy is commonly used to treat advanced prostate cancer. However, this treatment eventually becomes ineffective when cancer cells become androgen-independent. Interestingly, inhibiting AR expression even in this androgen-independent state results in inhibition of cell proliferation and cell death, suggesting that prostate cancer cells still depend on AR for survival. Only when the cellular mechanisms for this are better understood will novel therapeutics that target AR be able to reach their full potential.
Dr. Laura Lamb, recipient of a Fiscal Year 2007 Prostate Cancer Training Award (Predoctoral), has conducted studies to determine how AR and integrins, which are cell adhesion molecules that interact with the extracellular matrix (ECM), regulate survival in prostate cancer and normal prostate cells. To investigate the crosstalk between integrins, AR, and cancer cells interacting with the ECM, Dr. Lamb developed a new cell culture-based method in which prostate basal epithelial cells are induced to differentiate, after treatment with keratinocyte growth factor (KGF) and androgen, into a secondary layer of secretory epithelial cells. Her differentiation system was used to decipher the survival signaling pathways in these AR-expressing secretory cells that previously had been difficult to culture. Dr. Lamb demonstrated that survival of the secretory cells was dependent on E-cadherin and PI-3K, but not on androgen or AR. This indicated that survival was mediated via cell-cell interactions rather than through androgens. Although androgen was shown to be required for differentiation of basal epithelial cells into secretory cells, its role remains limited to the maintenance of androgen-dependent phenotype of the secretory cells (such as secretion of prostate-specific antigen) and is not required for secretory cell survival.
Dr. Lamb's research has led to one publication in the Journal of Cell Science, and she has submitted an application to patent her cell model, which will provide researchers with the opportunity to study specific mechanisms of survival in normal and cancerous cells. Ultimately, Dr. Lamb's cell model will facilitate the development of better treatments for prostate cancer.
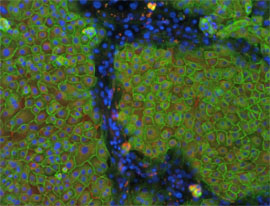
Prostate epithelial cell differentiation. Prostate epithelial cells were treated with KGF and androgen to induce differentiation. Prostate basal cells are positive for integrin beta 1 (green) and laminin 5 (red), while differentiated secretory cells are negative for both. Nuclei were visualized with Hoechst (blue). This model will be useful to investigate the cell biology of the prostate and prostate cancer (Adapted from Lamb et al., J. Cell Sci., 123:266-276, 2010).
Publication:
Lamb LE, Knudsen BS, and Miranti CK. 2010. E-cadherin-mediated survival of androgen receptor expressing secretory prostate epithelial cells derived from a stratified in vitro differentiation model. Journal of Cell Science 123:266-276.
Links: