



Posted April 10, 2014
Feng-Chun Yang, M.D., Ph.D., Indiana University, Indianapolis, Indiana
 Mutations in the NF1 tumor suppressor gene cause neurofibromatosis type 1 (NF1), a common genetic disorder. Individuals with NF1 are afflicted with a range of malignant and nonmalignant manifestations. Clinical studies have found that individuals with NF1 are at significant risk for both generalized and focal skeletal abnormalities including osteoporosis, dystrophic scoliosis, increased fracture rates, and failure of bone fractures to heal properly.
Mutations in the NF1 tumor suppressor gene cause neurofibromatosis type 1 (NF1), a common genetic disorder. Individuals with NF1 are afflicted with a range of malignant and nonmalignant manifestations. Clinical studies have found that individuals with NF1 are at significant risk for both generalized and focal skeletal abnormalities including osteoporosis, dystrophic scoliosis, increased fracture rates, and failure of bone fractures to heal properly.
Dr. Feng-Chun Yang of Indiana University has been the recipient of three separate awards to investigate the molecular mechanisms of bone abnormalities in NF1.
With funding from a Fiscal Year 2004 (FY04) New Investigator Award, Dr. Yang studied the biochemical and molecular mechanism underlying the pathological increase in de novo cytokine production in mouse Nf1+/- mast cells and human NF1 mast cells and evaluated whether mast cells from NF1 patients and mouse models have similar biological functions and cytokine secretion profiles. Dr. Yang and her research team showed that murine Nf1 +/- mast cells secreted elevated levels of TGF-ß, VEGF, and PDGF, which leads to the gain in fibroblast, endothelial cell, and Schwann cell biological functions. They also established that this gain in function is dependent on the Ras/PI3-K cell signaling pathway. In addition, they found that mast cells from NF1 patients have similar characteristics to those seen in the murine Nf1+/- mast cells, indicating that the biology of immune cells are comparable at certain levels between the human and murine system. This discovery is an important step in moving the field of NF1 research forward.
With funding from an FY07 New Investigator Award, Dr. Yang generated a mouse model to study the cellular and molecular mechanisms underlying the bone abnormalities in NF1. To investigate the role of Nf1 in the skeletal pathogenesis, Dr. Yang and her research team generated a conditional murine model in which one Nf1 allele was deleted from all tissues and the remaining allele was deleted only in osteoblasts. These mice showed increased development and activity of osteoclasts (cells that are specialized in bone resorption) and decreased numbers of osteoblasts (cells that are responsible for bone formation), which resulted in severe impairment of fracture healing in this mouse model. Interestingly, administration of the MEK inhibitor PD98059 significantly improved the fracture healing process, indicating that targeting the MAPK signaling pathways may improve bone formation and prevent bone resorption in NF1.
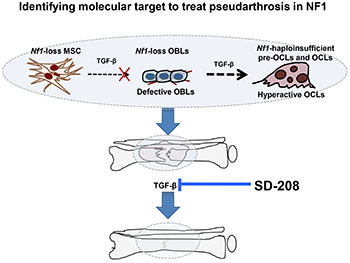
With funding from an FY10 Investigator-Initiated Focused Research Award, Dr. Yang and her research team sought to determine whether targeting the TGF-ß and MAPK pathway restores osteoclast functions, increases osteoblast differentiation, and thus enhances fracture healing in the NF1 mouse model. Using the mouse model above, which recapitulates skeletal abnormalities found in NF1 patients including low bone mass and tibial non-union fracture, Dr. Yang found significantly increased TGF-ß1 serum levels. The TGF-ß1 levels were also elevated in the serum of a cohort of NF1 patients. Further, she was able to demonstrate that inhibition of the TGF-ß signaling with the pharmacologic inhibitor of the TGF-ß receptor I kinase activity SD-208 rescued bone mass defects and prevented tibial fracture non-union in these mice. Overall, Dr. Yang's studies demonstrate that dysregulated TGF-ß 1 signaling is a primary factor underlying the pathogenesis of NF1-associated osteoporosis and non-union fracture. Therefore, pharmacologic inhibitors of TGF-ß signaling may be a potential therapeutic approach in the treatment for NF1 osseous defects, which are refractory to currently available treatments.
Several high-impact peer reviewed publications resulted from these funded studies, including "Hyperactive transforming growth factor-ß 1 signaling potentiates skeletal defects in a neurofibromatosis type 1 mouse model." Dr. Yang's student, Steven Rhodes, the first author of this publication, was the 2013 recipient of the Raisz-Drezner Journal of Bone and Mineral Research (JBMR) First Paper Award, which honors first authors of meritorious scientific publications published in the JBMR.
With the FY04, FY07, and FY10 NFRP funding, Dr. Yang and her research team have made significant progress in the field of NF1 research, which may lead to new therapeutic targets that will help alleviate some of the debilitating affects experienced by patients with NF1.
Publications:
Yang FC, Chen S, Robling AG, et al. 2006. Hyperactivation of p21ras and P13K cooperate to alter murine and human neurofibromatosis type 1-haploinsufficient osteoclast functions. J Clin Invest 116(11):2880-2891.
Wu X, Estwick SA, Chen S, et al. 2006 Neurofibromin plays a critical role in modulating osteoblast differentiation of mesenchymal stem/progenitor cells. Hum Mol Genet 15:2837-2845.
Yang FC, Chen S, Clegg T, et al. 2006. Nf1+/- mast cells induce neurofibroma like phenotypes through secreted TGF-b signaling. Hum Mol Genet 16:1-17.
Munchhof AM, Li F, White HA, et al. 2006. Neurofibroma-associated growth factors activate a distinct signaling network to alter the function of neurofibromin-deficient endothelial cells. Hum Mol Genet 15:1858-1869.
Nebesio TD, Ming W, Chen S, et al. 2006. Neurofibromin-deficient Schwann cells have increased lysophosphatidic acid dependent survival and migration-implications for increased neurofibroma formation during pregnancy. Glia 55:527-536.
Li F, Munchhof AM, White HA, et al. 2006. Neurofibromin is a novel regulator of RAS-induced signals in primary vascular smooth muscle cells. Hum Mol Genet 15:1921-1930.
Yan J, Chen S. Zhang Y, et al. 2008. Rac1 mediates the osteoclast gains-in-function induced by haploinsufficiency of Nf1. Hum Mol Genet 17(7):936-948.
Li H, Liu Y, Zhang Q, et al. 2009. Ras dependent paracrine secretion of osteopontin by Nf1+/- osteoblasts promotes osteoclast activation in a NF1 murine model. Pediatr Res 65:613-618.
Chen S, Burgin S, McDaniel A, et al. 2010. Nf1-/- Schwann cell-conditioned medium modulates mast cell degranulation by c-Kit-mediated hyperactivation of phosphatidylinositol 3-kinase. Am J Pathol 177:3125-3132.
Stevenson DA, Yan J, He Y, et al. 2011. Multiple increased osteoclast functions in individuals with NF 1. Am J Med Genet A 155A:1050-1059.
Zhang W, Rhodes SD, Zhao L, et al. 2011. Primary osteopathy of vertebrae in a neurofibromatosis type 1 murine model. Bone 48:1378-1387.
He Y, Staser K, Rhodes SD, et al. 2011. Erk1 positively regulates osteoclast differentiation and bone resorptive activity. PLoS One 6:e24780.
Wu X, Chen S, Orlando SA, et al. 2011. p85alpha regulates osteoblast differentiation by cross-talking with MAPK pathway. J Biol Chem 286:13512-13521.
Wu X, Chen S, He Y, et al. 2011. The haploinsufficient hematopoietic microenvironment is sufficient to normalize the pathological fracture repair in genetically engineered murine models of neurofibromatosis type 1. PLoS One 6(9):e24917.
He Y, Rhodes SD, Chen S, et al. 2012. c-Fms signaling mediates neurofibromatosis Type-1 osteoclast gain-in-functions. PLoS One 7(11):e46900.
Rhodes S, Wu X, He Y, et al. 2013. Hyperactive transforming growth factor-ß 1 signaling potentiates skeletal defects in a neurofibromatosis type 1 mouse model. J Bone Miner Res 28(12):2476-2489.
Sharma R, Wu X, Rhodes SD, et al. 2013. Hyperactive Ras/MAPK signaling is critical for tibial nonunion fracture in neurofibromin-deficient mice. Hum Mol Genet 1;22(23):4818-4828.
Links: