



MEK Inhibition Exhibits Efficacy in Human and Mouse Neurofibromatosis Tumors
Posted June 20, 2013
Nancy Ratner, Ph.D., Children's Hospital Medical Center, Cincinnati
Neurofibromatosis type 1 (NF1) results from mutations in the neurofibromin (NF1) gene, leading to alterations in intracellular signaling pathways that control cell growth. Children with NF1 are prone to developing benign tumors - called neurofibromas because they grow along the nervous system - that can cause disfigurement or disability and develop into lethal malignant tumors. These malignant tumors, known as peripheral nerve sheath tumors (MPNST), are resistant to conventional treatments and, consequently, there is an urgent need to develop new, more effective treatments for NF1 patients. Dr. Nancy Ratner, with funding from FY03 and FY08 Investigator-Initiated Research Awards from the Neurofibromatosis Research Program (NFRP), brought together a team of NF1 research experts to compare and contrast gene expression patterns associated with NF1 neurofibroma development and progression to malignant tumors, believing that gene profiles common to tumors from different patients would likely identify critical mechanisms of tumorigenesis and key therapeutic targets.
Using this approach, Dr. Ratner and colleagues recently identified a set of genes responsible for regulating the Ras/ERK signaling pathway that were activated in both benign neurofibromas and MPNSTs, suggesting that activation of this pathway is an early event in development of NF1 tumors. To determine if this pathway contributes to neurofibroma growth, Dr. Ratner then stained tissue sections using an antibody that recognizes the activated (phosphorylated) form of the ERK protein (P-ERK) as readout of Ras/ERK pathway activation. She found that activated ERK protein was increased in both NF1-related mouse and human tissue samples. P-ERK was elevated in NF1 nerve compared to wild type nerve, further elevated in neurofibroma, and most elevated in MPNST. These exciting data formed the basis for Dr. Ratner's decision to use PD0325901, a drug that inhibits one of the components of this pathway (MEK), in preclinical studies funded by Children's Tumor Foundation. She found that PD0325901 treatment stopped growth of neurofibroma and MPNSTs cells; dramatically shrank the size of both tumor types in most of the mice tested; and prolonged survival of mice implanted with malignant human NF1 tumors. These studies demonstrate that deregulated Ras/ERK signaling is critical for the growth of NF1 peripheral nerve tumors and provide a strong rationale for clinical trials of MEK inhibitors in children with neurofibromas.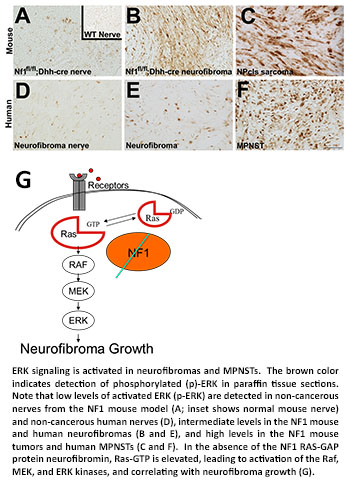
Publications:
Jessen WJ, Miller SJ, Jousma E, Wu J, Rizvi TA, Brundage ME, Eaves D, Widemann B, Kim MO, Dombi E, Sabo J, Hardiman Dudley A, Niwa-Kawakita M, Page GP, Giovannini M, Aronow BJ, Cripe TP, Ratner N. 2013. MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. Journal of Clinical Investigation 123(1):340-347.
Links: