 An official website of the United States government
An official website of the United States government
 ) or https:// means you've safely connected to the .mil website. Share sensitive information only on official, secure websites.
) or https:// means you've safely connected to the .mil website. Share sensitive information only on official, secure websites.
Parkinson's
Mechanisms of Specific Lipid-Induced Degeneration Causing Non-Motor Symptoms of Parkinson's Disease



Posted August 1, 2023
Ole Isacson, M.D., Ph.D., and Penelope Hallett, Ph.D., McLean Hospital

Ole Isacson, M.D., Ph.D.
(Photo Provided)
Penelope Hallett, Ph.D.
(Photo Provided)
Parkinson's disease (PD) is characterized by a range of motor symptoms, including slow movement, muscle stiffness and rigidity, and difficulty walking. In addition, approximately 80% of PD patients progress to dementia after the onset of motor impairment, making cognitive decline an important therapeutic target for the disease. A hallmark of PD is the formation of Lewy bodies, aggregates of fatty compounds called lipids coated with the synapse-associated protein α-synuclein (aSYN), in dopaminergic (i.e., dopamine-releasing) neurons of the striatum, the region of the brain that controls movement. The formation process of Lewy bodies, which ultimately lead to dopaminergic neuronal destruction, is still unclear, but there is growing evidence that lipid storage and transport abnormalities contribute to the pathology of PD and age-related dementias. An improved understanding of this dysfunction is a cornerstone of future therapeutic interventions. The Neurotoxin Exposure Treatment Parkinson's (NETP) program awarded Drs. Isacson and Hallett an Investigator-Initiated Research
In a normal, working central nervous system, the enzymes ß-hexosaminidase (HEX) and glucocerebrosidase (GCase) are important for the storage and removal of lipids within cell compartments called lysosomes. Importantly, the activity of both enzymes progressively decreases with age. Alterations of both proteins are suspected to contribute to Lewy body formation as deletion of the HEX gene produces aSYN aggregation in mice, and genetic mutations in GCase are a strong risk factor for PD in humans. As recently reported in Acta Neuropathologica Communications, Drs. Isacson and Hallett used an established lipid storage disorder animal model, HEX-deficient mice, to evaluate the role of HEX and aSYN in neuronal destruction. They found that loss of HEX activity in mice led to aSYN accumulation in the brain. Moreover, overexpression of HEX together with aSYN overexpression in the brains of rats, led to reduced accumulation of aSYN and prevented protein-lipid interactions by aSYN, as well as providing protection of midbrain dopamine-releasing neurons from aSYN-induced degeneration.
Having established that excess aSYN accumulates in lipid compartments, Drs. Isacson and Hallett and their team next explored the possibility that altered lipid trafficking plays a role in PD progression. In a 2020 Proceedings of the National Academy of Sciences article, the researchers analyzed total and cell-specific abundance of lipids in the striatum of postmortem brains from PD patients and healthy subjects. They found that, while the total abundance of neutral lipids (di- and triglycerides) in both the PD and non-PD striata was comparable, the distribution of neutral lipids across cell types shifted in PD patients: Elevated levels of neutral lipids and lipid inclusions, which colocalized with aSYN, in dopaminergic neurons from PD brains were countered by a proportional decrease in the level of neutral lipids found in neighboring astrocytes, which are brain cells that hold nerve cells in place. This inverse correlation is likely driven, at least in part, by the enzymatic activity of GCase, as the researchers observed that mice treated with GCase inhibitors not only accumulated aSYN aggregates but also exhibited a higher neutral lipid content in dopaminergic neurons and lower neutral lipid content in astrocytes compared to untreated mice.
While there are known genetic risk factors for PD, the disease also arises in people who have no genetic predisposition; these cases are called idiopathic (or sporadic) PD. Even though PD is primarily a neurological disease, genetic mutations and alterations to cellular mechanisms are present in all cells of the organism. The identification of systemic biomarkers of disease would enable early therapeutic intervention for patients with idiopathic PD. In their 2021 Molecular Brain research article, Drs. Isacson and Hallett generated an in-vitro cellular model of PD using skin fibroblasts from idiopathic PD patients and PD patients harboring the PD-associated N370S mutation in the gene encoding the GCase enzyme. Compared to fibroblasts from healthy controls, idiopathic or mutated PD fibroblasts exhibited decreased GCase activity. However, in contrast with genetic PD fibroblasts or fibroblasts from healthy subjects, idiopathic PD fibroblasts with reduced GCase activity also exhibited a decrease in the expression of lysosomal integral membrane protein 2 (LIMP2), a protein that regulates the activity and function of GCase. These results suggest that the reduction in GCase activity in idiopathic PD fibroblasts is driven by a different mechanism (alterations to LIMP2-mediated GCase activity) than that in genetic PD fibroblasts.
The work of Drs. Isacson and Hallett and their research team promises to provide clinicians with novel diagnostics, treatments, and therapeutics that will enable them to restore lipid equilibrium in the brains of PD patients by targeting glycolipid dysfunctions that play a role in triggering PD and related neurodegenerative diseases (as also outlined in their 2022 FEBS Journal review article). The development of such agents could significantly improve patient outcomes and quality of life.
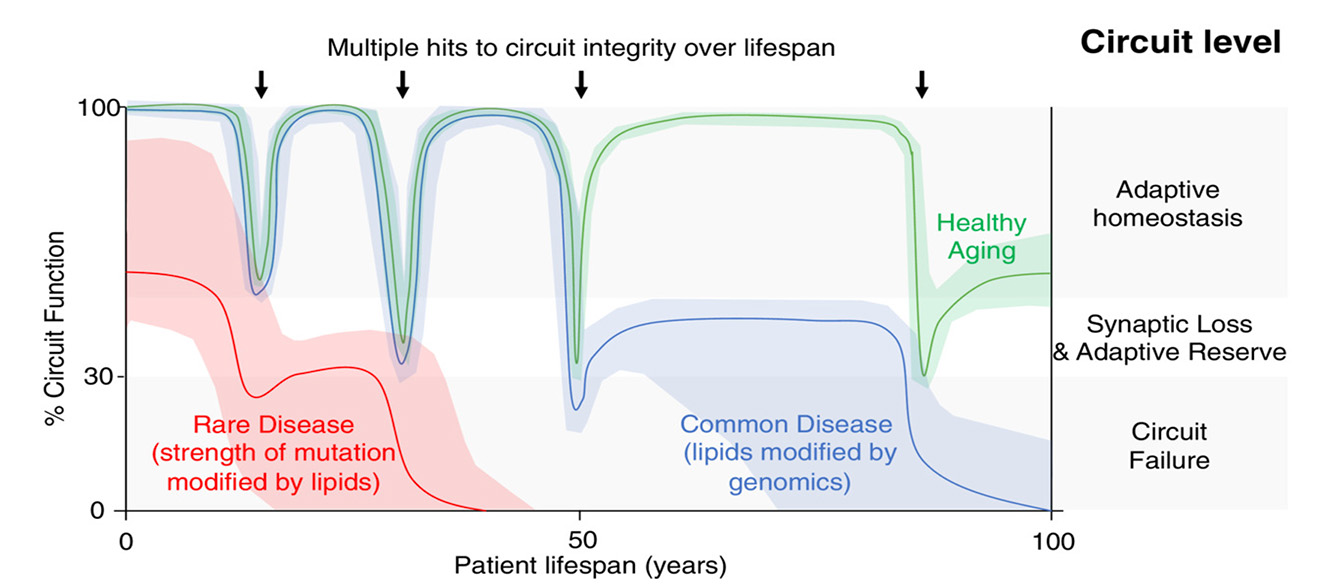 Figure 1. Repeated lifetime cellular lipid and metabolic stressors, in neural systems prone to Parkinson's, Lewy body dementia and Alzheimer's disease, may eventually lead to functional and degenerative failures (Cooper et al FEBS Journal 2022)
Figure 1. Repeated lifetime cellular lipid and metabolic stressors, in neural systems prone to Parkinson's, Lewy body dementia and Alzheimer's disease, may eventually lead to functional and degenerative failures (Cooper et al FEBS Journal 2022)
Publications:
Brekk OR, Honey JR, Lee S, et al. 2020. Cell type-specific lipid storage changes in Parkinson's disease patient brains are recapitulated by experimental glycolipid disturbance. Proceedings of the National Academy of Sciences U S A 117(44):27646-27654. https://doi.org/10.1073/pnas.2003021117
Brekk OR, Korecka JA, Crapart CC, et al. 2020. Upregulating β-hexosaminidase activity in rodents prevents α-synuclein lipid associations and protects dopaminergic neurons from α-synuclein-mediated neurotoxicity. Acta Neuropathologica Communications 8(1):127. https://doi.org/10.1186/s40478-020-01004-6
Cooper O, Hallett P, Isacson O. 2022. Upstream lipid and metabolic systems are potential causes of Alzheimer's disease, Parkinson's disease and dementias. FEBS Journal. https://doi.org/10.1111/febs.16638
Thomas R, Moloney EB, MacBain ZK, et al. 2021. Fibroblasts from idiopathic Parkinson's disease exhibit deficiency of lysosomal glucocerebrosidase activity associated with reduced levels of the trafficking receptor LIMP2. Molecular Brain 14(1):16. https://doi.org/10.1186/s13041-020-00712-3
Links:
Neuroregeneration Institute: www.neuroregeneration.org