
Previously Funded
Chronic Myelogenous Leukemia Research Highlights
2011:
2009:
- The Roles of SET and the Tumor Suppressor PP2A in CML Blast Crisis: Potential Therapeutic Targets
- Novel Inhibitors of Inosine Monophosphate Dehydrogenase
2008:
2007:
- Restoration of PP2A Tumor-Suppressor Activity as Therapeutic Strategy for Blast Crisis Chronic Myelogenous Leukemia
- The Fight Against CML Moves to the Garden
- Pyrrole-imidizole Polyamide Compounds Target CML Cells in a BCR-ABL-independent Manner
2006:
- Attacking Gleevec-Resistant CML Cells
- Combinations of Novel Histone Deacetylase and Bcr-Abl Inhibitors in the Therapy of Imatinib Mesylate-Sensitive and -Refractory Bcr-Abl Expressing Leukemia
- DNA Methylation May Impact Response to CML Therapy
2005:
- Advancing the Understanding of CML Progression
- Pre-Clinical Evaluation of PD166326, a Potential New CML Therapeutic Agent
2011
A Non-ATP Competitive Inhibitor of BCR-ABL for the Therapy of Imatinib-Resistant CMLs Posted April 26, 2011 E. Premkumar Reddy, Ph.D., Temple University, and
Ralph B. Arlinghaus, Ph.D., M. D. Anderson Cancer Center
Chronic myelogenous leukemia (CML) is a slowly progressing blood and bone marrow disorder characterized by the increased and unregulated clonal production of predominantly myeloid cells in the bone marrow and the accumulation of these cells in the blood. Approximately 4,870 new CML cases will be diagnosed in the United States in 2010. In almost all of these cases, CML is the result of a reciprocal chromosomal translocation identified as t(9;22). This translocation leads to production of the fusion protein BCR-ABL, which acts as a tyrosine kinase and is responsible for malignant transformation. The drug imatinib mesylate (IM; also known as Gleevec), an inhibitor of BCR-ABL, is effective in treating most CML patients. Unfortunately, relapse occurs in a subset of chronically treated patients and in those with advanced stages of CML due to mutations in the kinase domain of BCR-ABL, the target of IM.
Dr. Premkumar Reddy, recipient of a Fiscal Year 2005 Chronic Myelogenous Leukemia Research Program Therapeutic Development Award in collaboration with Dr. Ralph B. Arlinghaus, at M. D. Anderson Cancer Center, has focused on generating a potent inhibitor of BCR-ABL by targeting novel regions, thereby avoiding known IM-inactivating mutations.
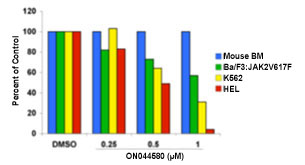
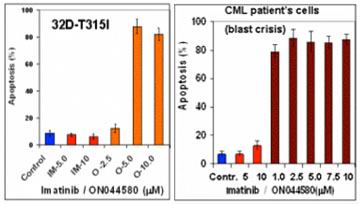
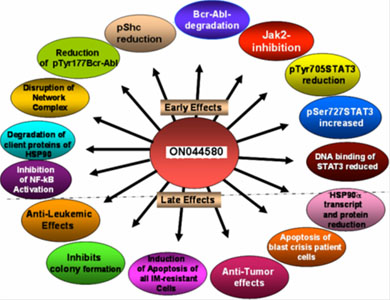
Using a rational drug design approach, a compound known as ON044580 was identified as the main candidate compound. The investigators found that ON044580 behaved as a dual protein kinase inhibitor, acting upon both BCR-ABL and Janus 2 kinase (JAK2) in vitro. ON044580 inhibited proliferation and induced apoptosis of leukemic cell lines expressing the V617F mutant form of JAK2 (Figure 1), which is associated with several myeloproliferative diseases (MPDs). In addition, ON044580 reduced the oncogenic behavior of cells expressing BCR-ABL and induced high levels of apoptosis in IM-resistant cells (Figure 2). These results are believed to be due to reduction of BCR-ABL protein levels and inhibition of Jak2 followed by the destruction of the BCR-ABL/JAK2/heat shock protein 90 (HSP90) signaling network complex (Figure 3). In further developmental work, Dr. Reddy has also established a nanoparticle strategy for the delivery of ON044580 into tumor cells. ON044580 is currently undergoing preclinical evaluation in preparation for Phase I human clinical trials.
Figure 1. Concentration-dependent growth inhibition of ON044580 on the three oncogenic cell lines (Ba/F3:jak2V617F, K562, and HEL cells), while it is not cytotoxic to normal mouse bone marrow cells.
Figure 2. ON044580-induced apoptosis in IM-resistant (32D-T315I) cells and cells from late stage of CML patients (blast crisis).
Figure 3. Summary of the multiple functions of ON044580 compound in BCR-ABL CML cells.
Study Publications:
Jatiani SS, Cosenza SC, Reddy MV, Ha JH, Baker SJ, Samanta AK, Olnes MJ, Pfannes L, Sloand EM, Arlinghaus RB, Reddy EP. 2010. A Non-ATP-Competitive Dual Inhibitor of JAK2 and BCR-ABL Kinases: Elucidation of a Novel Therapeutic Spectrum Based on Substrate Competitive Inhibition. Genes & Cancer 1(4):331-345.
Reddy MV, Pallela VR, Cosenza SC, Mallireddigari MR, Patti R, Bonagura M, Truongcao M, Akula B, Jatiani SS, Reddy EP. 2010. Design, Synthesis and Evaluation of (E)-Alpha-Benzylthio Chalcones as Novel Inhibitors of BCR-ABL Kinase. Bioorganic & Medicinal Chemistry 18(6):2317-2326.
Samanta AK, Chakraborty SN, Wang Y, Schlette E, Reddy EP, Arlinghaus RB. 2010. Destabilization of Bcr-Abl/Jak2 network by a Jak2/Abl kinase inhibitor ON044580 overcomes drug resistance in blast crisis chronic myelogenous leukemia (CML). Genes & Cancer 1:346-359.
2009
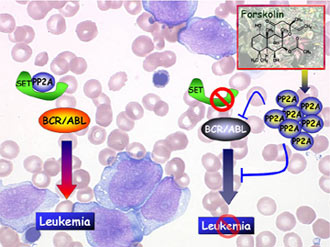
The Roles of SET and the Tumor Suppressor PP2A in CML Blast Crisis: Potential Therapeutic Targets Posted November 19, 2009 Danilo Perrotti, M.D., Ph.D., The Ohio State University, Columbus, Ohio
Chronic myelogenous leukemia (CML) is caused by a chromosomal translocation that creates a fusion oncoprotein called BCR-ABL. Unregulated BCR-ABL activity is thought to cause the expansion of pluripotent hematopoietic stem cells that leads to CML. Research efforts into BCR-ABL led to the development of imatinib, a therapeutic agent that targets BCR-ABL and is quite effective in treating CML patients in the stable phase of the disease. However, imatinib is not effective in managing CML during its acute phase, called blast crisis. Likewise, the recently discovered drug dasatinib is also not effective in the treatment of CML blast crisis. Researchers believe that the failure of these compounds to effectively treat the blast crisis stage is because CML-inducing stem cells persist under treatment. Therefore, many patients eventually progress to the blast crisis phase with limited treatment options. Thus, further investigation aimed at understanding the mechanisms by which the oncoprotein BCR-ABL abnormally regulates the growth of blood cells is required for the discovery of new potential therapeutic targets.
Through a fiscal year 2002 (FY02) Chronic Myelogenous Leukemia Research Program (CMLRP) Investigator-Initiated Research Award, Dr. Danilo Perrotti, of The Ohio State University, found that protein phosphatase 2A (PP2A), a tumor suppressor protein that exercises a critical role in cell growth, survival, and differentiation, is inhibited in BCR-ABL cells. This inhibition is due to BCR-ABL-induced overexpression of the leukemia-associated factor SET, a physiological PP2A inhibitor. Furthermore, Dr. Perrotti found that interference of the SET/PP2A interplay or pharmacologic PP2A activation suppresses BCR-ABL activity and expression, directly inactivating several important regulators of CML progenitor cell proliferation and survival.
These discoveries led Dr. Perrotti to propose the importance of PP2A inactivation for the pathophysiology of CML blast crisis and to generate preclinical data through a FY06 CMLRP New Investigator Award to further support the inclusion of PP2A-activating drugs in the therapy of imatinib-resistant and/or blast crisis CML.
Dr. Perrotti has since demonstrated that the drug FTY720, a known activator of PP2A, suppresses the growth and induces apoptosis of BCR-ABL-positive progenitor cells, but does not affect growth of normal human hematopoietic progenitor cells. Additionally, in a mouse model of CML, Dr. Perrotti found that FTY720 suppressed leukemia without inducing any toxic or adverse effects in animals. These results highlight the therapeutic relevance of rescuing PP2A tumor suppressor activity and strongly support the introduction of the PP2A activator FTY720 in the treatment of CML in the chronic phase and blast crisis.
In a collaborative mechanistic study, Dr. Perrotti found that inhibition of the Jak2 kinase, a downstream target of BCR-ABL, reduced the level of the SET protein in BCR-ABL cells. This led to increased activity of PP2A. Activation of PP2A combined with Jak2 inhibition enhanced the reduction of activated Lyn kinase, which has been associated with imatinib resistance. Inhibition of Jak2 induced apoptosis in CML cells from blast crisis patients, but not in normal hematopoietic cells. These results indicate that Lyn is downstream of Jak2, and Jak2 maintains activated Lyn kinase in CML through the SET-PP2A-Shp1 pathway. Dr. Perrotti has also found that PP2A activity might be essential for self-renewal of CML-initiating stem cells by inhibiting B-catenin-mediated transcription.
Therefore, activating PP2A during CML blast crisis and other hematologic malignancies might not only make the leukemic cells susceptible to tyrosine kinase inhibitors and other therapeutic agents, it may further inhibit production of new leukemic cells.
Publications:
Neviani P, Santhanam R, Trotta R, Notari M, Blaser BW, Liu S, Mao H, Chang JS, Galietta A, Uttam A, Roy DC, Valtieri M, Bruner-Klisovic R, Caligiuri MA, Bloomfield CD, Marcucci G, and Perrotti D. 2005. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell 8:355-368.
Perrotti D and Neviani P. 2006. ReSETting PP2A tumour suppressor activity in blast crisis and imatinib-resistant chronic myelogenous leukaemia. Br. J. Cancer 95:775-781.
Neviani P, Santhanam R, Oaks JJ, Eiring AM, Notari M, Blaser BW, Liu S, Trotta R, Muthusamy N, Gambacorti-Passerini C, Druker BJ, Cortes J, Marcucci G, Chen CS, Verrills NM, Roy DC, Caligiuri MA, Bloomfield CD, Byrd JC, and Perrotti D. 2007. FTY720, a new alternative for treating blast crisis chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphocytic leukemia. J Clin Invest 117(9):2408-2421.
Liu Q, Zhao X, Frissora F, Ma Y, Santhanam R, Jarjoura D, Lehman A, Perrotti D, Chen C-S, Dalton JT, Muthusamy N, and Byrd JC. 2008. FTY720 mediates cytotoxicity in chronic lymphocytic leukemia and lymphoblastic leukemia/lymphoma by a PP2A dependent mechanism. Blood 111(1):275-284 (Epub ahead of print Aug 2007).
Trotta R, Dal Col J, Allard J, Neviani P, Santhanam R, Mao H, Becknell B, Yu J, Modi A, Blaser BW, Perrotti D, and Caligiuri MA. 2007. The PP2A inhibitor SET regulates natural killer cells IFN-{gamma} production. J Exp. Med 204(10):2397-2405.
Perrotti D and Neviani P. 2008. Protein phosphatase 2A (PP2A), a drugable tumor suppressor in Ph1(+) leukemias. Cancer and Metastasis Reviews 27(2):159-168.
Samanta AK, Chakraborty SN, Wang Y, Kantarjian H, Sun X, Hood J, Perrotti D, and Arlinghaus RB. 2009. Jak2 inhibition deactivates Lyn kinase through the SET-PP2A-SHP1 pathway, causing apoptosis in drug-resistant cells from chronic myelogenous leukemia patients. Oncogene 28(14):1669-1681 (Epub 2009 Feb 23).
Roberts KG, Smith AM, McDougall F, Carpenter H, Perrotti D, Sim ATR, Ashman LK, and Verrills NM. 2009. Essential requirement for PP2A inhibition by c-KIT: Re-activation of PP2A as a treatment strategy for c-KIT+ cancers. Cancer Res (Submitted 2009).
Link:
Novel Inhibitors of Inosine Monophosphate Dehydrogenase Posted April 13, 2009 Krzysztof Pankiewicz, Ph.D., University of Minnesota, Minneapolis, Minnesota
Chronic myelogenous leukemia (CML), also known as chronic granulocytic leukemia or chronic myeloid leukemia, results from an overgrowth and accumulation of myeloid blood cells. Most CML cells carry the Philadelphia chromosome which results from a chromosomal translocation that creates a new gene, BCR-ABL, promoting unregulated cell growth and resulting in leukemia. CML cells also contain an isoform of inosine monophosphate dehydrogenase (IMPDH), type II that is expressed predominantly in cancer cells and activated blood cells. Type I of the IMPDH isoform is constitutively expressed in normal cells, and plays a key role in cell growth and development. Recently, type II IMPDH has emerged as a therapeutic target for the treatment of cancer and other diseases. Dr. Krzysztof Pankiewicz of the University of Minnesota was funded by the Department of Defense Chronic Myelogenous Research Program in fiscal year 2004 to develop inhibitors that are highly selective or specific against the type II isoform of IMPDH. Both IMPDH isoforms require a small cofactor molecule called nicotinamide adenine dinucleotide (NAD) for proper enzymatic activity. Tiazofurin is currently used as an orphan drug for the treatment of patients with CML. It is metabolized into tiazofurin adenine dinucleotide (TAD) which inhibits the natural cofactor NAD and thus inhibits IMPDH activity. However, CML patients often develop resistance to tiazofurin in which the drug is no longer effective at blocking IMPDH activity in CML cells. Dr. Pankiewicz and colleagues developed novel analogs of TAD, the downstream metabolic component of Tiazofurin, as well as other derivatives of NAD, the cofactor required for IMPDH activity, called mycophenolic adenine dinucleotides (MAD). Nearly 30 compounds were designed and evaluated for their ability to inhibit the type I and type II isoforms of IMPDH, and for their ability to inhibit cellular proliferation of the human CML cell line K562. Lead analogs were also evaluated for their anticancer activity in a SCID mouse model of CML. Some of the compounds showed great therapeutic potential, and Dr. Pankiewicz has opened new opportunities for the treatment of CML. A provisional patent has been filed regarding these novel potent inhibitors of IMPDH.
MAD analog docked to IMPDH2 Publications:
Chen L, Petrelli R, Olesiak M, et al. 2008. Bis(sulfonamide) isosters of mycophenolic adenine dinucleotide analogues: Inhibition of inosine monophosphate dehydrogenase. Bioorg Med Chem 16(15):7462-7469.
Chen L, Petrelli R, Felczak K, et al. 2008. Novel cofactor-type inhibitors of NAD-dependent enzymes: NAD-based therapeutics. Coll Czech Chem Commun Symp Series 10:71-79.
Chen L, Petrelli R, Felczak K, et al. 2008. Nicotinamide adenine dinucleotide (NAD) based therapeutics. Curr Med Chem 15:650-670.
Chen L, Gao G, Felczak K, et al. 2007. Probing binding requirements of type I and type II isoforms of IMP-dehydrogenase with adenine modified NAD analogues. J Med Chem 50:5743-5751.
Chen L, Gao G, Bonnac L, et al. 2007. Methylenebis-(sulfonamide) linked nicotinamide adenine dinucleotide analogue as an IMP-dehydrogenase inhibitor. Bioorg Med Chem Lett 17:3152-3155.
Bonnac LF, Gao G, Chen L, et al. 2007. Efficient synthesis of benzamide riboside, a potential anticancer agent. Nucelosides, Nucleotides & Nucleic Acids 26:1249-1253.
Rejman D, Olesiak M, Chen L, et al. 2006. Novel methylenephosphophosphonate analogues of mycophenolic adenine dinucleotide inhibition of inosine monophosphate dehydrogenase. J Med Chem 49:5018-5022.
Pankiewicz KW, Gao G, and Patterson SE. 2006. Synthesis of methylenebis(phosphonate) analogues of dinucleotide pyrophosphates and other P1, P2- methylenebis(phosphonate) diesters from methylenebis(phosphonate) monoesters. In: Current Protocols in Nucleic Acid Chemistry. Beaucage SL, Bergstrom DE, Herdewijn P, and Matsuda A, Eds. Wiley & Sons, Pages 13.5.1-13.5.13.
Link:
2008
Alternate paths towards a cure: The search for BCR-ABL kinase independent signaling in Chronic Myelogenous Leukemia Posted June 30, 2008Shaoguang Li, M.D., Ph.D., The Jackson Laboratory, Bar Harbor, Maine

The Philadelphia chromosome is created when the BCR and ABL genes located on human chromosomes 22 and 9, respectively, translocate to form the chimeric oncogene BCR-ABL. This oncogene constitutively expresses a tyrosine kinase that initiates the development of chronic myeloid leukemia (CML) as well as B-cell acute lymphoblasic leukemia (B-ALL). Currently, imatinib (Gleevec), a BCR-ABL tyrosine kinase inhibitor, is used to manage chronic phase CML, but is ineffective in blast phase CML or B-ALL. Additionally, imatinib cannot eliminate all BCR-ABL expressing cells and drug resistance can arise. In response to this problem, Dr. Shaoguang Li of The Jackson Laboratory, recipient of an FY05 Exploration Hypothesis Development Award, has sought to find BCR-ABL independent signaling pathways that play a role in the development of CML.
In this study, Dr. Li used fluorescence activated cell sorting (FACS) to analyze bone marrow (BM) cells transduced with a BCR-ABL retrovirus to discover the cell lineages functioning as CML stem cells. BCR-ABL expressing hematopoietic stem cells (HSCs) were identified as having a Lin-Sca-1+c-Kit+ profile. Experiments were conducted to determine the effects of the Src kinases Lyn, Hck and Fgr on BCR-ABL HSC renewal and survival in Src knockout mice using the drug dasatinib, a Src and BCR-ABL kinase inhibitor. Results in mice suggest that a lack of Src kinases prevents the transition of CML chronic phase to lymphoid blast crisis. Further experiments using imatinib and dasatinib in CML mouse models, including imatinib resistant mutant mice, were performed to determine if BCR-ABL-activated Src kinases confer imatinib insensitivity to acute lymphoblasic leukemia (ALL) in the CML blastic phase. Results demonstrated that Src kinases are responsible for ALL insensitivity to imatinib. Inhibition of Src kinases using dasatinib in B-ALL mice significantly prolonged their survival. Inhibition of BCR-ABL by imatinib did not have the same effect as dasatinib. Therefore, it was concluded that imatinib ineffectiveness in treating ALL mice was due to the inability of imatinib to inhibit Src kinase activity downstream of BCR-ABL signaling.
Despite the effectiveness of dasatinib in extending the lives of B-ALL mice, it was found that not all leukemic stems cells can be killed by BCR-ABL and Src kinase inhibition. In order to achieve long-term control over this disease, additional pathways need to be discovered and targeted.
Publications:
Hu Y, Swerdlow S, Duffy TM, et al. 2006. Targeting multiple kinase pathways in leukemic progenitors and stem cells is essential for improved treatment of Ph+ leukemia. Proc Natl Acad Sci USA 103(45):16870-16875.
Li S and Li D. 2007. Stem cell and kinase activity-independent pathway in resistance of leukemia to BCR-ABL kinase inhibitors. J Cell Mol Med 11(6):1251-1262.
Peng C, Brain J, Hu Y, et al. 2007. Inhibition of heat shock protein 90 prolongs survival of mice with BCR-ABL-T315I-induced leukemia and suppresses leukemic stem cells. Blood 110(2):678-685.
Link:
Abstract: A BCR-ABL Kinase Activity-Independent Signaling Pathway in Chronic Myelogenous Leukemia
2007
Restoration of PP2A Tumor-Suppressor Activity as Therapeutic Strategy for Blast Crisis Chronic Myelogenous Leukemia
Posted August 29, 2007
Danilo Perrotti, M.D., Ph.D., The Ohio State University, Columbus, OhioChronic myelogenous leukemia (CML) cells carry a chromosomal translocation that creates BCR/ABL, an oncogene with unregulated enzymatic activity. Initiation and progression of CML, a pluripotent hematopoietic stem cell disease, is attributed to BCR/ABL kinase activity that may be inhibited by therapeutic options such as imatinib mesylate and dasatinib. However, CML stem cells, which may play a role in disease recurrence, are not sensitive to treatment with BCR/ABL inhibitors. Furthermore, many CML patients develop resistance to these treatment options. Protein phosphatase 2A (PP2A), a protein that exercises a critical role in cell growth, survival, and differentiation, is reportedly downregulated in BCR/ABL-expressing cells. One downstream target of BCR/ABL kinase activity is SET, a naturally occurring inhibitor of PP2A that has been implicated in the negative control of cell growth and division, is significantly upregulated by BCR/ABL. Dr. Danilo Perrotti, of The Ohio State University, hypothesized that the inhibition of PP2A activity through increased BCR/ABL kinase activity occurs in the CML leukemia-initiating cell and plays an important role in maintenance and progression of this disease. Dr. Perrotti received a fiscal year 2006 Chronic Myelogenous Leukemia Research Program New Investigator Award to further the understanding of molecular mechanisms underlying development and progression of CML.
Dr. Perrotti and his colleagues used FTY720, a known immunomodulator, activator of PP2A and inhibitor of solid tumor growth, to study the effects of increasing PP2A activity in BCR/ABL-positive CML blast crisis (CML-BC) cells. The team demonstrated that FTY720 suppressed the growth and induced apoptosis of BCR/ABL positive myeloid and lymphoid progenitors in vitro but did not affect growth of normal human hematopoietic progenitor cells. Molecular studies revealed that FTY720 restored the activity of PP2A which, in turn, induced dephosphorylation of critical downstream BCR/ABL targets and suppressed BCR/ABL activity and expression. Not only did this group determine that FTY720 inhibited BCR/ABL leading to the inhibition of cell growth in CML-BC cells; they also determined that FTY720 inhibited the growth of BCR-ABL-positive CML cells that are imatinib mesylate- and dasatinib-resistant. Dr. Perrotti evaluated the effects of FTY720 on CML cells in vitro (in suspension culture and semisolid medium) and in a mouse model of CML, consistently finding that FTY720 inhibited growth of BCR/ABL positive cells without inducing any toxic or adverse effects in animals. These results indicate that the loss of PP2A activity in CML stem/progenitor cells may be critical to the development and recurrence of CML. Dr. Perrotti and colleagues suggest that incorporating PP2A-activating agents along with currently available therapeutic agents may improve the outcome for many patients.
Publication:
TNeviani P, Santhanam R, Oaks J, et al. 2007. FTY720, a new and alternative strategy for treating blast crisis CML and Ph1 ALL. Journal of Clinical Investigation (in press).
Link:
The Fight Against CML Moves to the Garden
Posted August 6, 2007
Craig Jordan, Ph.D., University of RochesterUnder normal circumstances, hematopoietic stem cells give rise to differentiated cells of the blood system. However, dysregulation of stem cell function may lead to the development of chronic myelogenous leukemia (CML). Recent research studies have demonstrated that CML stem cells (CML-SCs) may be resistant to currently available chemotherapeutic agents. The persistence of CML stem cells may play a role in recurrence of disease; therefore, CML-SCs represent an important potential target for the development of new therapeutic strategies. In a Fiscal Year 2002 Chronic Myelogenous Leukemia Research Program Investigator-Initiated Research Award titled "Targeted Ablation of CML Stem Cells," Dr. Craig Jordan proposed to (1) evaluate the role of NF-kB, which is upregulated in leukemic cells, in the biology of CML-SCs, and (2) investigate the conditions for inducing apoptosis in CML-SCs while sparing normal hematopoietic cells.
Studies designed by Dr. Jordan and his collaborator Dr. Katherine Borden addressing the role of NF-kB in the development of CML-SCs focused on the expression of eukaryotic translation initiation factor 4E (eIF4E), a NF-kB-regulated protein. Cellular levels of eIF4E affect the level of nucleocytoplasmic mRNA transport, and activation of NF-kB leads to increased eIF4E activity. Dr. Jordan's research demonstrated that elevated eIF4E expression levels were associated with suppressed differentiation of granulocytes and monocytes. Further studies determined that increased eIF4E levels caused a cascade of events leading to an increase in the transport of eIF4E-dependent mRNA molecules including transcripts for cyclin D, known to play an important role in the cell cycle. Analysis of primary CML cells demonstrated that the subcellular distribution of proteins such as cyclin D differed between primary leukemic and non-leukemic cells. Dr. Jordan and his colleagues used IkB, a repressor of NF-kB, to suppress NF-kB activity in cells. The subsequent reduction in NF-kB activity correlated with reduced expression of eIF4E and a decrease in eIF4E-dependent transcript transport suggesting that eIF4E-dependent mRNA transport may play a role in the development of cancer cells through its ability to affect cell growth and differentiation. Results from these studies implicate elevated NF-kB activity in leukemogenesis.
Dr. Jordan and his colleagues proposed that specifically targeting NF-kB activity may be a useful tool for targeting the destruction of CML-SCs. This research team designed studies using parthenolide (PTL), a naturally occurring small molecule found in the leaves of Tanacetum parthenium ("Feverfew"), an ornamental plant commonly displayed in gardens in the past and used for thousands of years to treat headaches. Parthenolide has been shown to inhibit NF-kB function, and recent medical history counts PTL as demonstrating antitumor activity through inhibiting DNA synthesis and cancer cell proliferation and sensitizing cells to other therapeutic agents. Dr. Jordan and his colleagues demonstrated that PTL induces apoptosis specifically in blast crisis CML (bcCML) cells and a leukemic stem cell (LSC) population. In short-term suspension cell culture, bcCML cells and LSC showed a strong cytotoxic response to PTL treatment whereas normal hematopoietic stem cells were significantly less affected. In vivo adoptive transfer studies demonstrated that engraftment of PTL-treated primary LSC was significantly reduced when compared to PTL-treated normal human stem cells. A screen of PTL-related compounds identified an analog with great promise. Dimethylamino-partenolide (DMAPT) was found to induce rapid death in human LSCs. The data from Dr. Jordan's research group demonstrate that the molecular mechanism of activity for PTL and DMAPT is through inhibition of NF-kB and changes in other cellular mechanisms and suggest that specifically targeting LSC using PTL or related analogs may result in the development of useful therapeutic agents for the treatment of CML.
Publications:
Topisirovic I, Guzman ML, McConnell MJ, et al. 2003. Aberrant eukaryotic translation initiation factor 4E-dependent mRNA transport impedes hematopoietic differentiation and contributes to leukemogenesis. Molecular and Cellular Biology 23(24):8992-9002.
Guzman ML, Rossi RM, Karnischky L, et al. 2005. The sesquiterpene lactone parthenolide induces apoptosis of human acute myelogenous leukemia stem and progenitor cells. Blood 105(11):4163-4169.
Link:
Pyrrole-imidizole Polyamide Compounds Target CML Cells in a BCR-ABL-independent Manner
Posted May 9, 2007
Joel Gottesfeld, Ph.D., The Scripps Research InstituteThe Chronic Myelogenous Leukemia Research Program granted a Fiscal Year 2004 Therapeutic Development Award to Dr. Joel Gottesfeld of The Scripps Research Institute for his research proposal, "Small Molecule Therapeutics for Chronic Myelogenous Leukemia." Dr. Gottesfeld's proposal is focused on developing a new class of small molecule therapeutic agents for the treatment of chronic myelogenous leukemia (CML). CML contains a genetic rearrangement of the Bcr and Abl genes resulting in the BCR-ABL fusion protein, a highly active tyrosine kinase (TK) believed to play a major role in the initiation and progression of CML. The development of TK inhibitors, such as imatinib mesylate, represented a major leap forward in the treatment of CML within the past decade; however, not all patients respond to this treatment and an increasing number of patients eventually develop resistance to it.
Dr. Gottesfeld identified small molecules, specific pyrrole-imidizole (Py-Im) polyamides, that inhibit gene-specific transcription. Py-Im polyamides have been shown to bind DNA sequences with high specificity and, when conjugated to DNA-modifying agents such as chlorambucil, can be used to inhibit DNA transcription. A screening study identified a Py-Im polyamide-chlorambucil conjugate, 1R-Chl, that downregulates the transcription of histone H4c (a key component of cellular chromatin), which is overexpressed in various cancer cells including CML. H4c represents a potential therapeutic target for CML. 1R-Chl specifically targets the DNA sequence 5'-WGGWGW-3' where W=A or T. In vitro 1R-Chl treatment of K562 cells, a human erythroleukemia cell line derived from a CML patient in blast crisis, resulted in a significant growth arrest of the cells in the G2/M phase of the cell cycle. In addition, data generated using a CML tumor xenograft murine model for CML (K562 cells) demonstrated that 1R-Chl significantly suppressed tumor growth in vivo without any apparent toxic effects.
An Affymetrix gene chip expression analysis revealed that 156 genes were influenced in 1R-Chl-treated K562 cells. Two of these genes, histones H4c and H4k/j, were of particular interest. A study of mRNA production demonstrated a decreased level of detectable H4c and H4k/j transcripts but not of Bcr-Abl gene transcripts in 1R-Chl-treated K562 cells and tumor cells derived from 1R-Chl-treated mice suggesting that the observed growth arrest is Bcr-Abl independent. While the mechanism of action for 1R-Chl is unclear, DNA damage is a likely suspect. Further studies of a synthesized small molecule library of related compounds identified another compound, 8R-Chl, which has greater antiproliferative properties and will be used in future studies.
Link:
Abstract: Small Molecule Therapeutics for Chronic Myelogenous Leukemia
2006
Attacking Gleevec-Resistant CML Cells
Posted October 16, 2006
Dr. E. Premkumar Reddy, Temple University
With increased knowledge of the basic biology of aberrant, disease-causing cells found in the body, therapeutics are now being designed to specifically target these cells while sparing normal cells. This holds true for chronic myelogenous leukemia (CML). Through the identification of a mutation in over 90% of CML patients that causes a mutated, constitutively active Bcr-Abl protein, researchers began focusing on inhibiting this protein. As a result, Gleevec® was created. This compound blocks the activity of Bcr-Abl by binding to the kinase domain of the protein, which is responsible for its activity in causing CML. However, following chronic treatment with Gleevec®, approximately 14% of patients develop a resistance to the drug through the development of mutations that prevents the binding of Gleevec® to Bcr-Abl. For this reason, researchers have been examining the Bcr-Abl molecule to find other ways to inhibit its activity. Dr. E. Premkumar Reddy at Temple University has identified several compounds that target a different area or conformation of Bcr-Abl than Gleevec®. Dr. Reddy believes that the region that these compounds bind to is less likely to undergo mutation. It was found that these compounds specifically inhibit Bcr-Abl and kill Gleevec-resistant tumor cells by apoptosis. Through a Department of Defense Chronic Myelogenous Leukemia Fiscal Year 2005 Therapeutic Development Award, Dr. Reddy is conducting further studies with these compounds to determine their precise mechanism(s) of action, pre-clinical toxicology, and pharmacological properties with the goal of initiating one or more clinical trials for treatment of CML patients.
Combinations of Novel Histone Deacetylase and Bcr-Abl Inhibitors in the Therapy of Imatinib Mesylate-Sensitive and -Refractory Bcr-Abl Expressing Leukemia
Posted September 28, 2006
Kapil Bhalla, M.D. Medical College of Georgia Cancer Center, Augusta, Georgia
The Bcr-Abl tyrosine kinase, a key factor in the developmenet of chronic myelogenous leukemia (CML), is a protein that can have a dramatic effect on the growth and survival of CML cells in the body. In non-CML cells, Bcr-Abl is not expressed. However, in CML, the unchecked activity of Bcr-Abl results in leukemic transformation of cells. For this reason, Bcr-Abl is an important therapeutic target in the treatment of CML, as exemplified through the use of imatinib mesylate (Gleevec®). By targeting the deregulated Bcr-Abl tyrosine kinase, Gleevec® is highly active in inducing clinical and cytogenetic remission. However, studies of patients who relapse demonstrate that after 42 months of treatment, approximately 16% of patients develop mutations, making them resistant to Gleevec®.
Dr. Kapil Bhalla of the Medical College of Georgia Cancer Center is addressing this problem with funding from a Department of Defense Chronic Myelogenous Leukemia Research Program Fiscal Year 2004 Therapeutic Development Award. Dr. Bhalla hypothesizes that use of histone deacetylase (HDAC) inhibitors in combination with Bcr-Abl inhibitors, such as Gleevec®, may circumvent the resistance-associated mutations. HDACs function to remove the acetyl group chemical modification from proteins; the addition or removal of this group can change a protein's behavior. Previously, Dr. Bhalla found that inhibition of HDAC-6 led to an increase in the acetylated form of heat shock protein 90 (hsp90). This acetylation inhibits the normal chaperone function of hsp90, which typically maintains the proper function and activity of a number of proteins in the cell. Among those proteins chaperoned by hsp90 is Bcr-Abl. Dr. Bhalla has now found that the loss of hsp90 function following treatment with HDAC-6 inhibitors leads to depletion of Bcr-Abl in both Gleevec®-sensitive and -resistant primary CML cells, causing cell death. Furthermore, by combining the HDAC inhibitor with nilotinib or dasatinib, both potent next-generation Bcr-Abl inhibitors, an additive effect was found whereby Gleevec®-resistant blast crisis cells were reduced to a greater extent than with either treatment alone. This is a promising new approach to treatment that may overcome resistance by targeting Bcr-Abl from several directions.
Publications:
Bali P, Pranpat M, Bradner J, et al. 2005. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: A novel basis of antileukemia activity of histone deacetylase inhibitors. J Biol Chem 280:26729-26734.
Fiskus W, Pranpat M, Bali P, et al. 2006. Combined effects of novel tyrosine kinase inhibitor AMN107 and histone deacetylase inhibitor LBH589 against Bcr-Abl expressing human leukemia cells. Blood 108:645-652.
Fiskus W, Pranpat M, Balasis M, et al. Co-treatment with vorinostat (suberoylanilide hydroxamic acid, SAHA) enhances activity of dasatinib (BMS-354825) against imatinib mesylate sensitive or resistant chronic myelogenous leukemia cells. Clin Cancer Res (in press).
DNA Methylation May Impact Response to CML Therapy
Posted May 15, 2006
Jean-Pierre J. Issa, University of Texas M.D. Anderson Cancer Center
Chronic myelogenous leukemia (CML) is often treated successfully with imatinib mesylate, a tyrosine kinase inhibitor. However, resistance to imatinib develops in about 15% of patients and is often mediated through mutations in BCR/ABL. The development of resistance may correlate with stage of disease - early chronic phase to late chronic phase (CP); accelerated phase (AP); and blastic phase (BP). Increased epigenetic silencing of potential tumor suppressor genes correlates with disease progression, which suggests a relationship between epigenetic silencing of genes with imatinib resistance. Dr. Jean-Pierre Issa received a Department of Defense Chronic Myelogenous Leukemia Research Program Investigator-Initiated Research Award in fiscal year 2002 for his proposal titled "Epigenetic silencing and resistance to imatinib mesylate in CML." Dr. Issa and his colleagues hypothesized that methylation-associated epigenetic silencing in gene promoter regions contributes to clinical resistance to imatinib treatment. Understanding the level of correlation may help to determine which patients may not respond well to imatinib mesylate treatment and may aid in the development of new treatments and regimens.
DNA samples from 200 patients diagnosed with CML were used to determine genes that were potentially informative for DNA methylation in CML patients. Genes identified include: P15, PGRA, PGRB, CDH13, NOR1, NPM2, DPYS, and RIL. Data generated from this study indicate that DNA methylation not only increases with disease progression but this increase is more robust in patients who are resistant to imatinib mesylate. Dr. Issa then hypothesized that imatinib mesylate-resistant CML cells would respond to treatment with 5-aza-deoxycytidine (decitabine) because decitabine, an inhibitor of DNA methyltransferases, had previously been demonstrated to cause a hypomethylation status in treated cells. He also hypothesized that imatinib sensitivity would be restored in decitabine-treated cells. A Phase II clinical trial was conducted to determine the effect of decitabine treatment on DNA methylation status and to evaluate clinical response in imatinib-resistant CML patients. Decitabine was administered by IV over several cycles. Hypomethylation status of peripheral blood mononuclear cells derived from decitabine-treated, imatinib-resistant CML patients in CP, AP, and BP was evaluated using a bisulfite/pyrosequencing assay. The data demonstrated that decitabine was active in imatinib-resistant CML and that methylation was suppressed in decitabine-treated cells.
The relationship between methylation status of cells and the patient's response to decitabine treatment was evaluated. Methylation status before administration of cycle 1 treatment did not apparently correlate with response to treatment. However, analyses of hypomethylation status after the end of therapy showed that the level of DNA methylation was greatly decreased in those patients who did not respond to decitabine treatment when compared to patients who responded to treatment. Issa and colleagues suggested that this result is due to an inherent ability of therapy-resistant cells to withstand increased levels of hypomethylation that normally subject therapy-sensitive cells to death.
Publication:
Issa JPJ, Gharibyan V, Cortes J, et al. 2005. Phase II study of low-dose decitabine in patients with chronic myelogenous leukemia resistant to imatinib mesylate. Journal of Clinical Oncology 23:3948-3956.
Link:
Abstract: Epigenetic Silencing and Resistance to Imatinib Mesylate in CML
2005
Advancing the Understanding of CML Progression
Posted November 17, 2005
Danilo Perrotti, Ph.D., Ohio State UniversityUnderlying mechanisms of chronic myelogenous leukemia (CML) progression from the chronic phase to the blast crisis stage are not well understood and probably involve changes in a range of cellular and molecular processes. For example, changes in the regulation of mRNA metabolism may correlate with CML progression. Expression levels of RNA-binding proteins hnRNP K and hnRNP La, involved in the regulation of mRNA translation, are increased in BCR/ABL-expressing myeloid cell lines and in primary cells from CML patients in blast crisis stage. Danilo Perrotti, Ph.D., of Ohio State University, designed a research program focused on evaluating the role that active BCR/ABL protein, an oncogenic tyrosine kinase found in CML blast crisis patients, plays in hnRNP K- and La-dependent translation regulation that might account for phenotypic changes. In Fiscal Year 2002, Dr. Perrotti received a Department of Defense Chronic Myelogenous Leukemia Research Program Investigator-Initiated Award for a research proposal titled "Effect of BCR/ABL Oncoprotein on mRNA Translation: Possible Mechanisms Responsible for CML Disease Progression."
Using a CML-derived, BCR/ABL-expressing cell line, K562, the Principal Investigator proposed to use Ribonomics to identify hnRNP K- and hnRNP La-associated mRNAs that may contribute to the blast crisis CML cell phenotype. Both hnRNP La and hnRNP K RNA-binding proteins were found to be strongly associated with the mRNA of the tumor suppressor protein phosphatase 2A (PP2A), a serine/threonine phosphatase that is made up of three subunits (catalytic subunit, structural subunit, substrate specificity subunit). Expression of PP2A activity is inhibited in CML patients who have progressed to blast crisis stage. The Ribonomics screen demonstrated that hnRNP La was associated with the mRNA of PP2A regulatory subunit B, alpha (PPP2R3A), suggesting that expression of this subunit is regulated during CML blast crisis and may be associated with the suppression of PP2A activity in CML patients. PP2A activity may also be regulated by the expression of a physiologic inhibitor of PP2A called SET. Expression of SET is enhanced by increased expression of BCR/ABL and is increased during progression of CML. Dr. Perrotti and colleagues demonstrated that another RNA-binding protein, hnRNP A1, in BCR/ABL-positive hematopoietic cells may associate with the mRNA that encodes for SET. Treatment of BCR/ABL-positive cells with suppressed PP2A activity with imatinib, which blocks BCR/ABL kinase activity, reestablished PP2A activity. Dr. Perrotti and his colleagues demonstrated that expression of active PP2A, either through molecular or pharmacologic means, lead to the dephosphorylation, and therefore inactivation, of regulators that are important for the proliferation and survival of CML progenitor cells. These data suggest that new CML treatments may be possible through the development of therapeutics aimed at upregulating PP2A activity in BCR/ABL-positive CML.
Pre-Clinical Evaluation of PD166326, a Potential New CML Therapeutic Agent
Posted June 14, 2005
Robert L. Ilaria Jr., M.D., University of Texas Southwestern Medical CenterMost chronic myelogenous leukemia (CML) arises as a result of a reciprocal chromosomal translocation event causing the expression of a fusion protein, Bcr/Abl, atyrosine kinase with potent leukemogenic activity. Imatinib mesylate (Gleevec, STI 571) has successfully treated CML patients in the early stages of disease; however, treatment of patients in the later stages of CML has not been as successful, and there is evidence of emerging imatinib mesylate-resistant CML. The development of alternative treatment options such as the compound PD166326 might benefit CML patients. Robert L. Ilaria Jr., M.D. received a Department of Defense Chronic Myelogenous Leukemia Research Program Investigator-Initiated Award for a proposal titled "Novel Targeted Approaches for de Novo and ST1571 (Imatinib)-Resistant Bcr/Abl-induced Leukemia" for research designed to study PD166326 in a pre-clinical mouse model of human CML. PD166326 is a promising potential CML therapeutic agent. Using this mouse model, Dr. Ilaria and his colleagues produced in vivo data that was the first to move studies of PD166326 from the in vitro setting to the in vivo setting, a critical first step that will bring this agent closer to testing in human clinical trials. Dr. Ilaria and his colleagues demonstrated that PD166326 (1) suppressed leukemic cell growth, (2) suppressed tyrosine phosphorylation, (3) was well tolerated, (4) quickly reached therapeutic levels in the blood stream, and (5) was useful in treating an imatinib-resistant CML mouse model. PD166326 treatment of a mouse model with a CML-like myeloproliferative disorder reduced leukemic cells better than treatment with imatinib mesylate. Additionally, splenomegaly, a characteristic of this CML-like myeloproliferative disorder, was reduced along with peripheral blood granulocytosis when compared to imatinib mesylate- or placebo-treated mice. PD166326 also suppressed tyrosine phosphorylation events. PD166326 and imatinib mesylate suppressed overall tyrosine phosphorylation when compared to placebo-treated mice; global suppression of constitutive phosphotyrosine was greater with PD166326. Specifically, the phosphorylation status of Crk-L, a target of Bcr/Abl kinase activity, and Lyn, a Src family member implicated in the development of imatinib mesylate-resistant CML, was suppressed in PD166326-treated mice. To address the issue of emerging imatinib mesylate-resistant CML, Dr. Ilaria and colleagues studied the effects of PD166326 using a mouse model of imatinib-resistant CML in which the bone marrow was reconstituted with cells expressing mutants of Bcr/Abl implicated in imatinib-resistant CML. PD166326-treated mice reconstituted with P210/H396P mutant protein-expressing bone marrow cells survived twice as long as placebo-treated mice. These data taken together suggest that PD166326 may be an effective addition to the arsenal of CML treatment options in the future.
Publications:
Wolff NC, Veach DR, Tong WP, Bommann WG, Clarkson B, and Ilaria RL. 2005. PD166326, a novel tyrosine kinase inhibitor, has greater antileukemic activity than imatinib mesylate in a murine model of chronic myeloid leukemia. Blood 105:3995-4003.
Last updated Friday, July 29, 2022