- Deciphering the Vital Role of Skp2 in the Progression and Metastasis of Prostate Cancer
- Revealing Vitamin D Pathways to Prostate Cancer Prevention and Treatment
- Stem Cell Therapy Shows Promise for Preventing Metastatic Prostate Cancer
- Discovery of New Drug Candidates for the Prevention of Hormone Refractory Prostate Cancer
- Androgen Receptor Synergizes with Increased Androgen to Drive Gene Fusion in Prostate Cancer Cells
- Key Discovery to Interfere with Lethal Prostate Cancer
- PCRP-Funded Researchers are Revealing the Link between Cholesterol-Lowering Drugs and the Potential Prevention of Advanced Prostate Cancer
- Identifying a Key Culprit in Prostate Cancer Metastasis
- Improving Biopsy Techniques in Recurrent Prostate Cancer
- Sunitinib Advances to a Phase III Clinical Trial in Men with Advanced Prostate Cancer
Deciphering the Vital Role of Skp2 in the Progression and Metastasis of Prostate Cancer
Posted December 16, 2010
Hui-Kuan Lin, Ph.D., University of Texas M. D. Anderson Cancer Center, Houston, Texas
Developing more effective therapies to treat advanced prostate cancer is a critical component to saving lives. PCRP investigators are striving to better understand the mechanisms underlying aggressive disease to identify molecular targets against which therapies can be developed. Dr. Hui-Kuan Lin, a Fiscal Year 2008 New Investigator Award recipient, and colleagues have identified a protein called Skp2 (S-phase associated protein 2) that is often overexpressed in prostate cancer cells, but whose inactivation causes tumor cells to lose their ability to divide (and therefore multiply), a phenomenon known as cellular senescence. Novel therapeutics that induce senescence of tumor cells while leaving normal cells untouched represent a promising new approach for preventing prostate cancer progression.
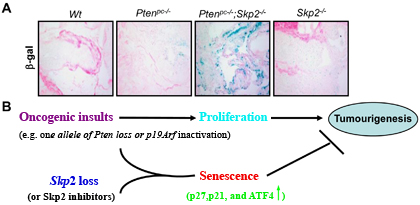
The potential of Skp2 inactivation was discovered through it's interference with the effects of inactivating another gene known as PTEN. One of the most common genetic alterations in prostate cancer is the deletion of the PTEN tumor suppressor gene, which occurs in approximately 40% of prostate cancer patients. It was previously shown that inactivation of the PTEN gene in the prostate glands of mice results in spontaneous development of invasive prostate cancer. Dr. Lin developed a mouse model in which both Skp2 and PTEN are inactivated in the prostate gland and found that tumor size and weight in these mice were decreased by almost 80% compared to mice with only PTEN inactivation. Histological studies of the prostate glands from these mice showed that loss of Skp2 in PTEN-inactivated cancer cells decreases cellular proliferation, increases cellular senescence, and inhibits development of invasive prostate cancer. These results suggest that Skp2 may be a novel therapeutic target for clinical intervention in prostate cancer progression and metastasis.
Furthermore, using a small molecule inhibitor (MLN4924) that blocks Skp2's involvement in marking proteins for degradation, Dr. Lin demonstrated in cell culture that interfering with the downstream activity of Skp2 induces cellular senescence in PTEN-inactivated cancer cells and inhibits tumor cell growth in xenograft models of human prostate cancer. Together, the data indicate an important role for Skp2 in the regulation of cell cycle arrest and senescence, and corroborate that pharmacological inactivation of Skp2 represents a promising new approach for the treatment of advanced prostate cancer.
Skp2deficiency restricts prostate cancer development by triggering cellular senescence. A) Senescence analysis of anterior prostate from Ptenpc-/- and Ptenpc-/- Skp2-/- mice aged 15 months. B) Working model for tumour-suppressive cellular senescence driven by oncogenic insults and Skp2deficiency (Adapted from From Lin et al., Nature, 464, 374-379, 2010).
Publication:
Lin HK, Chen Z, Wang G, Nardella C, Lee SW, Chan CH, Yang WL, Wang J, Egia A, Nakayama KI, Cordon-Cardo C, Teruya-Feldstein J, and Pandolfi PP. 2010. Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature 464(7287):374-379.
Chan CH, Lee SW, Li CF, Wang J, Yang WL, Wu CY, Wu J, Nakayama KI, Kang HY, Huang HY, Hung MC, Pandolfi PP, and Lin HK. 2010. Deciphering the transcriptional complex critical for RhoA gene expression and cancer metastasis. Nature Cell Biology 12(5):457-467.
Links:
Revealing Vitamin D Pathways to Prostate Cancer Prevention and Treatment
Posted September 14, 2010
Nancy L. Weigel, Ph.D., Baylor College of Medicine, Houston, Texas
Supported by Fiscal Year 2003 (FY03) and FY08 PCRP Idea Development Awards, Dr. Nancy Weigel is discovering the mechanisms by which vitamin D, well-recognized to inhibit growth of prostate cancer cells in culture and in some animal tumor models, can best be exploited for prostate cancer prevention and treatment.
Higher exposure to sunlight (the major contributor to vitamin D production in the body) correlates with decreased risk for prostate cancer. Phase II/III clinical trials are already in progress to test the potential of vitamin D and its derivatives to prevent or delay the progression of early- and late-stage prostate cancer. Although the precise mechanisms through which vitamin D inhibits prostate cancer growth are still unclear, it has been shown that the active metabolite of vitamin D, 1,25-dihydroxyvitamin D3 (1,25D), and a highly potent analog, EB1089 (seocalcitol), bind the vitamin D receptor (VDR) in cells and subsequently turn on the expression of genes that inhibit prostate cancer cell growth. A better understanding of specific changes in gene expression and their role in prostate cancer development will provide insight toward maximizing the efficacy of vitamin D in preventing and treating the disease.
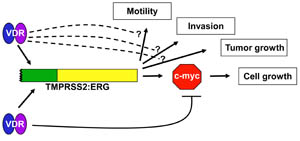
With her PCRP funding, Dr. Weigel sought to specifically identify the genes regulated by vitamin D in both normal and cancerous prostate cells. One of the genes whose expression was suppressed in non-transformed (RWPE-1), androgen-dependent (LNCaP and LAPC-4), and androgen-independent (C4-2) prostate cancer cells was c-Myc, an oncogenic transcription factor frequently overexpressed in prostate cancer. Using small inhibitory RNA (siRNA) knockdown experiments of c-Myc expression in C4-2 cells, Dr. Weigel showed that the reduction in c-Myc expression by siRNA treatment was similar to that observed with 1,25D treatment. In both 1,25D and siRNA-treated cells, there was a comparable decrease in cell proliferation. Using androgen-dependent prostate cancer cells (LNCaP and LAPC-4) treated with or without EB1089, she showed that about 25 genes were upregulated and 7 genes were downregulated by EB1089 in both cell lines. One of the upregulated genes was TMPRSS2. As this androgen regulated gene plays a role in the TMPRSS2-ETS gene fusions found in over 50% of prostate tumors and is associated with more aggressive disease, Dr. Weigel examined the effect of 1,25D on the TMPRSS2-ERG fusion gene present in the androgen-dependent prostate cancer cell line VCaP. She found that 1,25D and EB1089 both increased the expression of the TMPRSS2-ERG fusion gene, but reduced the growth and proliferation of VCaP cells. Furthermore, vitamin D decreased the expression of c-Myc, a TMPRSS2-ERG target gene and transcription factor that is overexpressed in a variety of cancers and is a critical component for cell proliferation. Thus, 1,25D may be beneficial in prostate cancers that express the TMPRSS2-ETS gene fusions, but additional studies including studies in vivo are needed.
Dr. Weigel, a dedicated teacher who has also received FY05 and FY08 PCRP Collaborative Undergraduate Historically Black Colleges and Universities Student Summer Training Program Awards, directs the Prairie View A & M/Baylor College of Medicine SMART Summer Undergraduate Prostate Cancer Research Project, which is a 9-week summer prostate cancer research experience designed to encourage minority college students to pursue careers in biomedical science.
The vitamin D receptor (VDR) induces TMPRSS2:ERG expression, but down-regulates c-Myc, an ERG target. The TMPRSS2:ETS factor fusions, present in greater than 50% of prostate cancers, stimulate motility, invasiveness, and cell and tumor growth. Dr. Weigel's research team's finding that VDR induces TMPRSS2 led them to test the response of VCaP cells expressing the TMPRSS2:ERG fusion to VDR activation. Treatment with the active metabolite of vitamin D, 1,25-dihydroxyvitamin D3, induced TMPRSS2:ERG, but reduced cell growth and expression of c-Myc, an ERG target gene in this cell line. Whether there are additional actions of 1,25-dihydroxyvitamin D3 that counteract ERG stimulated motility, invasiveness, and growth of tumors in vivo remains to be determined.
Publication:
Rohan JN and Weigel NL. 2009. 1alpha,25-dihydroxyvitamin D3 reduces c-Myc expression, inhibiting proliferation and causing G1 accumulation in C4-2 prostate cancer cells. Endocrinology 150(5):2046-54.
Washington MC and Weigel NL. 2010. 1alpha,25-dihydroxyvitamin D3 inhibits growth of VCaP prostate cancer cells despite inducing the growth promoting TMPRSS2:ERG gene fusion. Endocrinology 151(4):1409-1417.
Links:
Public and Technical Abstracts: TMPRSS2-ERG Gene Fusions and Vitamin D Action in Prostate Cancer
Stem Cell Therapy Shows Promise for Preventing Metastatic Prostate Cancer
Posted September 14, 2010
Selvarangan Ponnazhagan, Ph.D., University of Alabama at Birmingham
Bone metastasis is a major factor contributing to prostate cancer mortality and morbidity, causing severe bone pain and fractures. Because conventional chemotherapy and radiation therapy do not provide long-term protection from prostate cancer bone metastases, new therapies for the clinical control of bone metastases and the resulting bone damage are needed. Dr. Selvarangan Ponnazhagan of the University of Alabama, in results published in the journal Clinical Cancer Research, has recently shown that adult mesenchymal stem cells have therapeutic potential to inhibit the growth of prostate tumors in the bone and prevent bone damage.
Bone is a dynamic tissue that is continually being remodeled by the action of osteoclasts, which break down bone, and osteoblasts, which build bone. In order for prostate cancer cells to form bone metastases, osteoclasts must first degrade the bone at the metastatic site to establish a niche for cancer cells to seed and grow. This process can be inhibited by blocking the action of key regulators such as a protein called RANKL (receptor activator of NF-kB ligand), which is produced by bone marrow stromal cells, binds to receptors on osteoclasts, and stimulates bone resorption activity of osteoclasts. Denosumab, a monoclonal antibody that targets the RANKL protein, has recently completed Phase III clinical trials in patients undergoing androgen deprivation therapy. Another key regulator is osteoprotegerin (OPG) which is secreted by osteoblasts and functions as a "decoy receptor" by competing with RANK for RANKL binding, and thus preventing osteoclast-induced bone resorption and metastasis.
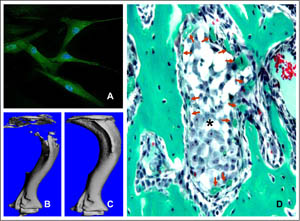
Since bone marrow-derived adult mesenchymal stem cells (MSCs) can differentiate into osteoblasts and produce osteoprotegerin, Dr. Ponnazhagan, recipient of an FY05 PCRP Idea Development Award proposed to study the potential of using OPG-secreting MSCs to decrease bone damage and prevent prostate cancer bone metastasis. His group implanted tumor-forming human prostate cancer cells (PC-3) into the tibia of mice, and after 24 hours, injected MSCs that were either unmodified or genetically modified to overexpress osteoprotegerin. Four weeks after treatment, mice were analyzed by live imaging, and the data showed that the MSCs caused a 90% decrease in tumor growth in both treatment groups, showing that the genetically modified MSCs had no advantage over unmodified cells, and demonstrating that MSC therapy can prevent prostate cancer growth in bone by minimizing the area for tumor growth through effective bone remodeling. However, in similar experiments in mice with tumors that had been allowed to grow for 2 weeks before treatment (i.e. established bone tumors showing a high degree of bone damage), the treatment failed to show a significant advantage. Taken together, the results show that MSCs can provide therapeutic benefit during the early lesion phase but are not effective after the bone tumor metastasis has established itself and the bone has been severely damaged. Dr. Ponnazhagan concluded that MSCs delivered at an early stage of the disease may have the potential to provide a therapeutic advantage in limiting the establishment of prostate cancer bone metastases. In addition, he suggests that since MSCs in the bone already exist in relatively low quantities, strategies to stimulate their growth and proliferation in bone metastases could provide novel new therapy to reduce morbidity and mortality for prostate cancer bone lesions.
Therapeutic potential of MSCs in osteolytic bone metastasis. (A) Immunostaining indicated murine and human MSCs constitutively express osteoprotegerin (OPG) in culture (green fluorescence). (B) Growth of bone metastatic prostate cancer cells (PC3) in the mouse tibia generates severe osteolytic lesions. (C) MSCs expressing OPG prevents bone loss via inhibition of osteoclastogenesis, which is required by the tumor cells for growth in bone. (D) In addition, MSCs formed woven bone around tumor nests (*), which further reduced tumor progression. Bone sections were stained with Goldner's trichrome stain, and collagen is stained green.
Publication:
Chanda D, Isayeva T, Kumar S, Hensel JA, Sawant A, Ramaswamy G, Siegal GP, Beatty MS, Ponnazhagan S. 2009. Therapeutic potential of adult bone marrow-derived mesenchymal stem cells in prostate cancer bone metastasis. Clinical Cancer Research 15(23):7175-7185.
Links:
Discovery of New Drug Candidates for the Prevention of Hormone Refractory Prostate Cancer
Posted August 24, 2010
Marianne Sadar, Ph.D., British Columbia Cancer Agency, Vancouver, British Columbia, Canada
Dr. Marianne Sadar of the British Columbia Cancer Agency has recently isolated and characterized a novel small molecule inhibitor of the androgen receptor (AR) that blocks the growth of castration-recurrent prostate cancer (CRPC), previously called hormone refractory or androgen-independent. The results, published in Cancer Cell, show that, unlike current anti-androgen therapies in clinical trials, this new inhibitor targets the amino-terminal region of the AR and prevents protein-protein interactions that drive the gene expression that leads to uncontrolled growth of CRPC. These exciting results hold promise for the development of an effective therapeutic for aggressive, late-stage prostate cancer.
Radical prostatectomy and radiation therapy are effective treatments for primary prostate cancer, but some patients develop recurrent tumors that are usually treated with androgen deprivation therapy. Androgen deprivation is initially effective, causing blood prostate specific antigen (PSA) levels and tumor burden to decrease. Over time, however, the tumors recur in a castration resistant form. The current treatment for CRPC is docetaxel combined with prednisone, which on average increases survival by only a few months. Failure of this treatment to improve long-term survival contributes to CRPC's role in the estimated 32,000 deaths from prostate cancer this year. Clearly, new therapeutic approaches are needed if we are to reduce or eliminate prostate cancer deaths.
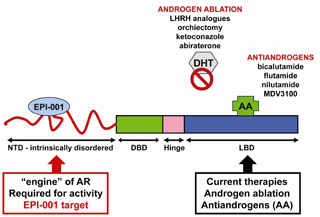
The AR is a protein composed of distinct structural domains including one that binds androgen (ligand-binding domain [LBD]), another that binds to specific DNA sequences (DNA-binding domain [DBD]), and an amino-terminal domain (NTD) that activates transcription. The AR becomes activated in the cytoplasm by binding with androgens (testosterone and dihydroxytestosterone), then moves to the nucleus where it turns on the expression of genes that cause prostate cancer cells to grow, and turns off genes that cause them to die. Binding of androgen to the LBD of the AR results in its activation, enabling the receptor to bind to specific DNA sequences, termed androgen response elements (ARE), in androgen-regulated genes such as PSA. Anti-androgen therapies used to treat prostate cancer, such as bicalutamide, interfere with the binding of androgen to the LBD and are effective in androgen-dependent but not CRPC. Since the AR assumes a nuclear (active) rather than cytoplasmic (inactive) location, and many of the same genes turned on or off by androgen and AR in androgen-dependent prostate cancer are also on or off in castration-resistant tumors, then it is likely that the AR is functioning in CRPC in the absence of testicular androgen.
Dr. Sadar, recipient of Fiscal Year 2004 (FY04) and FY06 Idea Development Awards, used high-throughput screening techniques to search a library of over 1700 marine sponge extracts for compounds that would inhibit AR activity in both androgen-dependent and -independent prostate cancer cells. One promising extract, EPI-001, was selected and further characterized. Dr. Sadar showed that EPI-001 binds to the AR NTD, blocks AR-induced gene expression regardless of whether androgens were present, and inhibits the proliferation of prostate cancer cells expressing the AR (LNCaP, MDA PCa2B, and 22RV1), but not the proliferation of prostate cancer cells lacking AR (DU145 and PC3). To determine if EPI-001 could inhibit the growth of prostate tumors, Dr. Sadar used a mouse model of both androgen-dependent and androgen-independent prostate cancer (LNCaP). AR-positive prostate tumors were allowed to grow to 100 mm3 in male mice and then treated with EPI-001 by intravenous injection. After 20 days, the tumors in untreated and non-castrated mice had grown to a large size (400 mm3) whereas the tumors in EPI-100 treated mice were much smaller (200 mm3). To determine if EPI-001 would prevent the growth of CRPC, the tumors were again allowed to grow to 100 mm3 and then the mice were deprived of androgen by castration. After 2 weeks, the tumors in untreated mice grew to 148 mm3 whereas in the EPI-001 treated mice, the tumor had decreased in size from 100 mm3 to 73 mm3 less than half that of untreated mice. Similar experiments using intra-tumoral rather than intravenous injection showed a 12-fold decrease in size by 25 days after beginning EPI-001 treatment. The results show that EPI-001 is non-toxic, slows the growth of both androgen-dependent and CRPC tumors, and may actually shrink tumors of lethal CRPC. Since currently there are no effective therapies for advanced stage prostate cancer, EPI-001 targeting the NTD of the AR holds promise for the development of an effective therapeutic for lethal prostate cancer.
The N-terminal domain (NTD) of the androgen receptor is a therapeutic target. EPI-001 inhibits the NTD of the androgen receptor, which is required for its activity. Currently, all other therapies such as androgen ablation and antiandrogens (AA) target the ligand-binding domain (LBD). [DNA binding domain (DBD); dihydrotestosterone (DHT)]
Publication:
Anderson RJ, Mawji NR, Wang J, et al. 2010. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell 17:535-546.
Links:
Public and Technical Abstracts: Novel Approaches for Blocking Activation of the Androgen Receptor
Androgen Receptor Synergizes with Increased Androgen to Drive Gene Fusion in Prostate Cancer Cells
Posted August 6, 2010
Michael Rosenfeld, M. D., University of California, San Diego, La Jolla, California
Dr. Michael Rosenfeld, of the University of California, San Diego, has recently published exciting new findings that may help explain the origin of somatic gene fusions that occur frequently in prostate tumors. The results, published in the journal Cell, describe a model in which androgen receptor (AR) is recruited to sites near common chromosomal breakpoints, inducing structural changes that bring the TMPRSS2 gene in proximity to ETS family genes and enable their fusion. TMPRSS2-ETS gene fusions are found in over 50% of patients with prostate cancer and may provide a key to developing therapeutics, biomarkers, and greater insight into the origins and progression of the disease.
Gene fusions between the regulatory region of TMPRSS2 (an androgen-responsive gene) and ETS family genes, most commonly ERG (ETS related gene) or ETV1 (Ets variant gene 1), by chromosomal translocation, result in these genes being controlled by androgens and the AR and increase the expression of ETS genes in some tumors. These fusions have been hypothesized to lead to the development of more aggressive prostate cancer. Previous studies suggested that TMPRRS2 gene fusions occur randomly and that their increased frequency was the result of selection of subpopulations of tumor cells that had acquired a growth advantage via increased levels of ETS gene products. Dr. Rosenfeld, with grant support from a Fiscal Year 2007 Prostate Cancer Research Program Idea Development Award, found that TMPRSS2, ERG, and ETV gene fusions in prostate cancer are not random events but, rather, are orchestrated by AR, thus establishing a key mechanism for how these genetic alterations arise in prostate cancer.
Because of the AR's critical role in prostate cancer, and the ability of radiation to rapidly induce chromosomal translocations, Dr. Rosenfeld examined the effects of androgens and radiation in a cellular model of androgen-responsive prostate cancer in which TMPRSS2-ETS translocations are not present. He observed that adding androgens or exposing the cells to radiation induced the formation of TMPRSS2-ETS gene fusions. Fusion events with each treatment occurred at a low frequency (3% to 5% of the samples); however, application of androgens and radiation in combination induced higher levels of fusion transcripts (30% to 35% of the samples). DNA damaging agents such as etoposide or doxorubicin similarly synergized with androgen to increase the frequency of gene fusions. Analysis of DNA and fusion transcripts from treated cells confirmed the presence of chromosomal breakpoints and fusion junctions similar to those found in tumors from prostate cancer patients. Interestingly, in the presence of androgen, AR was recruited to binding sites near the breakpoints and induced conformational changes in the chromosomes that brought the TMPRSS2 gene close to ERG and ETV1 genes, suggesting a mechanistic "smoking gun" for fusion events.
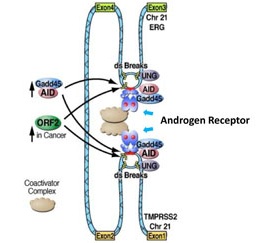
Similar experiments performed in normal prostate epithelial cells did not result in gene fusions, suggesting that additional factors are present in prostate cancer cells that may promote the induction of gene fusions. Dr. Rosenfeld identified two genes that may be responsible for this key difference: Activation-induced deaminase (AID) and Line-1 retroelement ORF2 endonuclease (ORF2). Although not normally present in the prostate gland, these genes were found to be expressed in prostate cancer and recruited by AR to breakpoint regions. Both genes have previously been associated with the introduction of DNA breaks, suggesting that they may be critical to the formation of the gene fusions found in prostate cancer. Together, the new findings suggest a model to explain how TMPRSS2-ETS gene fusions arise. The breakage and fusion seems to be orchestrated by AR via its binding sites near the breakpoint regions. Understanding the molecular mechanisms underlying the formation of TMPRSS2-ETS gene fusions will not only tell us a great deal about prostate cancer progression, but new discoveries in this emerging field may be essential keys to development of effective new diagnostics, prognostics, and therapeutics for prostate cancer.
A schematic illustration of androgen receptor-mediated chromosomal translocations leading to formation of TMPRSS2-ETS gene fusions. The androgen receptor with bound androgen brings the TMPRSS2 and ERG genes (normally located at a distance from each other on chromosome 21 [light blue string]) close together near the sites of double-strand breaks (ds Breaks) resulting in fusion of the TMPRSS2 and ERG genes. High levels of activation-induced deaminase (AID) and Line-1 retroelement ORF2 endonuclease (ORF2) expressed in prostate cancer cells (but not normal prostate cells) are brought to DNA breakage sites by androgen receptor along with Gadd45 (a transcriptional activator), uracil-DNA glycosylase (UNG), and coactivator proteins (Coactivator Complex) to facilitate formation of the gene fusion.
Publication:
Lin CL, Yang B, Tanasa K, Hutt BG, Ohgi JK, Zhang J, Rose DW, Fu XD, Glass CK, and Rosenfeld MG. 2009. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell 139(6):1069-1083.
Links:
Key Discovery to Interfere with Lethal Prostate Cancer
Posted June 4, 2010
Michael Karin, Ph.D., University of California-San Diego, San Diego, California
A key factor involved in the failure of androgen deprivation therapy has been identified in a mouse model of prostate cancer. Inflammation triggered by dying tumor cells was shown to activate a signaling pathway that prompts androgen-independent growth of prostate cancer cells, which may provide new targets for treatment of the most lethal form of the disease, androgen-independent/castration-resistant prostate cancer.
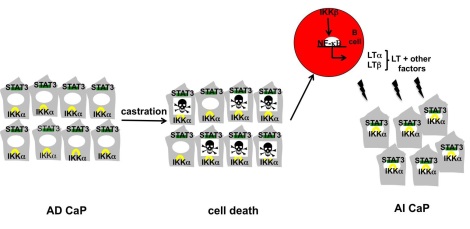
Radical prostatectomy and/or radiation therapy are two effective modalities often used for treating primary prostate cancer. However, despite their effectiveness, in some patients there is a recurrence of tumors. These recurrent tumors are androgen-dependent, prompting a second line of treatment using androgen deprivation (or ablation) therapy. Androgen deprivation causes hormone-dependent tumors to shrink, but over time the tumors emerge in an androgen-independent form capable of growing in a low-androgen environment. These androgen-independent/castration-resistant tumors are usually more aggressive and more metastatic than the original tumor. There are currently no effective therapies for androgen-independent prostate cancer, and the reasons underlying the eventual failure of androgen deprivation therapy are poorly understood. Dr. Michael Karin, recipient of a Fiscal Year 2003 Prostate Cancer Research Program Idea Development Award, has identified an inflammatory response triggered by dying prostate cancer cells inside tumors. The dying tumor cells recruit B cells (white blood cells), which infiltrate the tumor and produce the protein lymphotoxin, an inflammatory cytokine. Lymphotoxin induces the proliferation of residual prostate cancer cells, resulting in tumor growth in the absence of androgens.
Using a mouse model of prostate cancer, Dr. Karin allowed tumors to grow to a large size (1000 mm3), after which the mice were androgen deprived by castration. He observed that prostate cancer cells began to die, reducing the tumors dramatically to about 50 mm3, suggesting that androgen deprivation can effectively kill tumor cells in mice. However, tumors re-emerged after two weeks and increased to the original size (1000 mm3) within four weeks of androgen-deprivation therapy. Dr. Karin found that B cells, derived from the bone marrow and not present in the initial androgen-dependent tumors, had infiltrated the androgen-independent tumors, suggesting that the development of androgen-independent prostate cancer was dependent on an inflammatory/immune response mediated by B cells. He showed that, as androgen-starved tumors began to die, they secreted a protein called CXCL13, whose function is to attract B cells to the sites of tissue injury, in this case the dying prostate tumors. As the B cells infiltrate these tumors they secrete lymphotoxin. Lymphotoxin interacts with a receptor on the surface of prostate cancer cells, called lymphotoxin beta receptor, and activates cell signaling to NF-KB, a protein that controls DNA transcription. This in turn stimulates tumor cell proliferation in the absence of androgen stimulation. Taken together, these results suggest that the inflammatory response to dying tumor cells contributes to the eventual failure of androgen deprivation therapy. Further investigation by Dr. Karin showed that neither antibodies that block CXCL13 nor inhibitors of the NF-KB signaling had an effect on androgen-dependent tumor cells. However, both treatments were shown to delay the emergence of androgen-independent prostate cancer by 3-4 weeks after castration (androgen depletion). These results suggest that therapeutic interventions that prevent lymphotoxin production and/or signaling during androgen deprivation therapy could delay the emergence of androgen-independent prostate cancer in humans by up to 2-3 years.
A model explaining how therapy-induced inflammation promotes emergence of androgen-independent prostate cancer. Androgen withdrawal causes death of primary androgen-dependent (AD) prostate cancer cells (CaP), which release mediators that lead to recruitment of inflammatory/immune cells, including B cells, into the tumor remnant. Activation of IKB kinase(IKK) bin B cells results in NF-KB-dependent production of lymphotoxinand other factors that activate IKKa and STAT3 in residual prostate cancer cells. This results in enhanced cell survival, proliferation and growth of androgen-independent (AI) prostate cancer.
Publication:
Ammirante M, Luo JL, Grivennikov S, Nedospasov S, Karin M. 2010. B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature 11;464(7286):302-5.
Links:
PCRP-Funded Researchers are Revealing the Link between Cholesterol-Lowering Drugs and the Potential Prevention of Advanced Prostate Cancer
Posted April 26, 2010
Lionel Bañez, M.D., Duke University, Durham, North Carolina
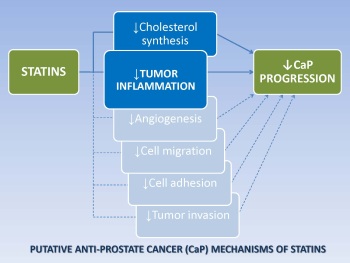
Statins, commonly used in the treatment of heart disease, are effective agents for lowering cholesterol levels in the blood. Recent studies suggest that statins may also reduce inflammation. This may be a major benefit, as inflammation is associated with advanced prostate cancer, and increasing evidence suggests that statin use may reduce the risk of advanced prostate cancer. Dr. Lionel Bañez, recipient of Fiscal Year 2006 (FY06) Health Disparity Training-Prostate Scholar Award and FY08 Health Disparity Research Award (Transitioning Investigator Category), considered the link between statins and reduced inflammation and hypothesized that statin use might slow or prevent the progression of prostate cancer to advanced disease by decreasing inflammation within prostate tumors.
To test this hypothesis, Dr. Bañez examined prostate cancer tissues from 236 radical prostatectomy patients, 37 (16%) of whom had used statins for a period of 1 year before surgery. The degree of inflammation in prostate cancer tissues was determined based on the number of inflammatory cells present in the tumors. He found that, in general, inflammatory cells were present in 82% of the prostate tumors and, among these, 36% of the tumors had high numbers of inflammatory cells. Comparison of prostate tumors from statin users and nonusers showed that the tumors from patients who had used statins had lower numbers of inflammatory cells. Dr. Bañez concluded that statin use was significantly associated with lower risk for prostate tumor inflammation, and the lower risk was even more pronounced in those patients who had used high doses of statins.
This is the first study to show an association between preoperative statin use and a reduction of intratumoral inflammation in prostate cancer patients. Although these findings require confirmation in larger clinical studies, the work suggests that statins reduce the risk of developing advanced prostate cancer by preventing inflammation in the tumor.
Publication:
Bañez LL, Klink JC, Jayachandran J, Lark AL, Gerber L, Hamilton RJ, Masko EM, Vollmer RT, Freedland SJ. 2010. Association between statins and prostate tumor inflammatory infiltrate in men undergoing radical prostatectomy. Cancer Epidemiol Biomarkers Prev 19(3),722–728.
Links:
Identifying a Key Culprit in Prostate Cancer Metastasis
Posted March 31, 2010
Karen Cichowski, Brigham and Women's Hospital, Boston, Massachusetts
The majority of the 27,000 deaths caused by prostate cancer each year result from the spread (metastasis) of prostate cancer cells to other parts of the body. There are currently no effective therapies for patients with metastatic prostate cancer. Although much is known about the genetic changes and the intracellular signaling pathways controlling prostate cancer cell growth, very little is known about these processes as they relate to prostate cancer metastasis. Dr. Karen Cichowski, a Fiscal Year 2007 PCRP Idea Development Awardee, demonstrated that DAB2IP (“disabled homolog 2 interacting protein”) is turned off by the process of epigenetic silencing in prostate tumors by a second gene, EZH2, which is upregulated in prostate cancer progression. Of crucial importance, she showed that the gene DAB2IP controls metastatic spread of prostate cancer by coordinately regulating two distinct cell signaling pathways.
To understand the function of DAB2IP in the development of prostate cancer metastasis, Dr. Cichowski used cell models of normal prostate (PrEC) and prostate cancer (PC-3) and found that, consistent with the data from prostate tissues, DAB2IP is expressed in normal cells but is silenced in prostate cancer cells. Normally, PC-3 cells produce metastatic tumors when injected into mice; however when Dr. Cichowski activated expression of the silenced DAB2IP gene in the PC-3 cells, the cells were no longer able to produce tumors in mice. To verify the results, a DAB2IP-specific short hairpin RNA (shRNA) was used to specifically turn off DAB2IP expression in PrEC cells, and when these cells were injected into mice, high-grade invasive and metastatic tumors formed. Similarly, introduction of EZH2 into normal PrEC cells resulted in the silencing of DAB2IP, and mice injected with these cells also developed invasive and metastatic prostate cancer. Dr. Cichowski also demonstrated that silencing of DAB2IP in normal PrEC cells activated both the Ras oncogene pathway and the NF-kB signaling pathway. She found that the Ras oncogene pathway induced uncontrolled cell proliferation, and the activated NF-kB signaling pathway was critical for metastatic spread of prostate cancer. EZH2, therefore, activates the Ras and NF-êB pathways by epigenetically suppressing DAB2IP, providing the molecular mechanism by which an epigenetic regulator activates these two major signaling pathways. Taken together with the data from human tissues, these results suggest a critical role for DAB2IP in suppressing prostate cancer tumor development and metastasis, and reveal EZH2 and DAB2IP as potential biomarkers for progression to metastasis and new therapeutic targets for reducing death and suffering due to prostate cancer.
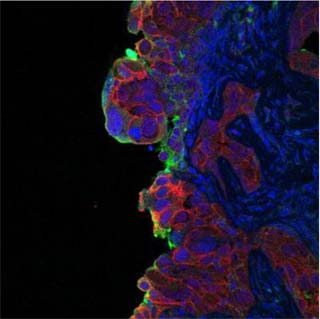
DAB2IP loss induces prostate cancer metastasis by causing dramatic molecular changes at the invasive front of the tumor. This image shows a DAB2IP-deficient metastatic tumor in mice undergoing an “epithelial to mesenchymal”-like transition at the leading edge of the tumor. Green: the mesenchymalmarker vimentin. Red: the epithelial marker e-cadherin. Blue: DAPI staining of cell nuclei.
Publication:
Min J, Zaslavsky A, Fedele G, McLaughlin SK, Reczek EE, De Raedt T, Guney I, Strochlic DE, Macconaill LE, Beroukhim R, Bronson RT, Ryeom S, Hahn WC, Loda M, Cichowski K. 2010. An oncogene-tumor suppressor cascade drives metastatic prostate cancer by coordinately activating Ras and nuclear factor-B. Nature Medicine 16 (3):286-294.
Links:
Improving Biopsy Techniques in Recurrent Prostate Cancer
Posted March 4, 2010
Cynthia Ménard, M.D., University Health Network, Toronto, Ontario, Canada
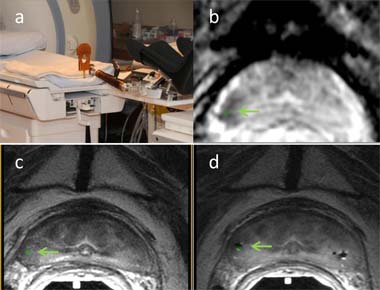
For some men, radiation therapy as treatment for early stage prostate cancer is only partially effective, risking continued tumor growth and/or metastasis. Dr. Cynthia Ménard, a Fiscal Year 2005 Prostate Cancer Research Program New Investigator Awardee, investigated the combined use of Magnetic Resonance Imaging (MRI) and prostate needle biopsies to better identify and target any remaining cancerous tissue for salvage therapy. A special MRI table was designed to allow needle placement while the patient was lying on his back (rather than side or stomach) to improve prostate gland stability during the procedure. This table was tested in conjunction with integrated diagnostic and interventional MRI using stereotactic trans-perineal needle navigation for real-time imaging of needle placement. MRI-guided biopsies were performed on thirteen prostate cancer patients with suspected growing tumors who had previously received radiation therapy (median 5.8 years prior), and the procedure was well tolerated by these patients with minimal discomfort.
Using MRI measurements and biopsy results, Dr. Ménard created three-dimensional tumor maps of the prostate gland to correlate diagnostic MRI data with biopsy tissue histology for identification of recurring tumor sites. Ten patients were found to have recurrent disease, with the site of recurrence corresponding to suspicious imaging findings in all but two cases, demonstrating that MRI can spatially delineate local prostate cancer recurrence after radiation treatment. Precise focusing of additional treatment after radiotherapy to areas of recurrent tumor growth within the patientÂ’s prostate gland may help prevent cancer spread, which in turn may translate into improved cure rates with fewer side effects for prostate cancer patients.
In additional ongoing clinical studies funded by this award, Dr. Ménard is using MRI to measure oxygen levels in growing tumors since it is widely accepted that tumors containing low oxygen levels tend to respond differently to therapy. By measuring oxygen in tumors and comparing the results to MRI, physicians may be able to choose and adapt salvage therapies according to the level of oxygen in the tumor in different parts of the prostate gland, further improving prostate cancer cure rates.
MRI-Guided Biopsy. (a) Dedicated interventional MRI table assembly providing pelvic access for stereotactic needle guidance during integrated diagnostic imaging and biopsy. (b) Navigation software (Aegis, Sentinelle Medical, Inc.) displaying needle target highlighted in green (see light green arrow pointing to green target area) onto suspicious dark tumor visible on ADC map. (c) Corresponding anatomic T2 weighted images. (d) Needle verification images documenting actual location of 3 biopsy needles (signal voids R and L) in reference to intended suspicious tumor target highlighted in green. Biopsy confirmed the presence of malignancy.
Links:
Sunitinib Advances to a Phase III Clinical Trial in Men with Advanced Prostate Cancer
Posted January 27, 2010
M. Dror Michaelson, M.D., Ph.D., Massachusetts General Hospital, Boston, Massachusetts
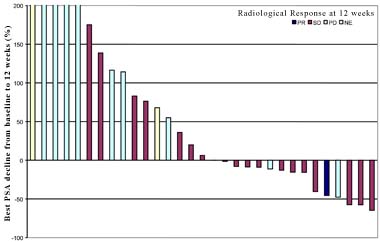
A drug already in use for treatment of some types of cancer has now been shown to have potential for treating the lethal form of prostate cancer as well. Advanced metastatic castration-resistant prostate cancer (CRPC) develops in a subset of prostate cancer patients and causes an estimated 27,000 deaths per year in the United States. There is an urgent need to develop improved treatments for CRPC. Sunitinib (distributed as Sutent by Pfizer) is a drug that targets growth factor receptors such as those for vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF). These receptors are expressed on the surface of endothelial and prostate cancer cells and, upon binding their respective growth factors, activate intracellular pathways controlling cell proliferation. Since inhibition of VEGF slows the growth of CRPC in vitro and in vivo, Dr. Michaelson conducted a Phase II clinical trial of sunitinib in two groups of men with CRPC. Thirty-four patients enrolled in the trial were treated for 12 weeks with sunitinib. Before starting treatment, all patients had rising prostate specific antigen (PSA) values or worsening bone scans; some patients had previously received docetaxel-based chemotherapy. Overall, 4 patients showed declines in PSA value, although this was transient for 2 of the patients, and 3 patients showed radiographic improvements.
Although the treatment was well-tolerated, data on PSA failed to show significant advantage. However, in clinical studies of advanced prostate cancer, increasing evidence suggests that PSA response may not be an adequate indicator of anti-tumor activity. Therefore Dr. Michaelson examined angiogenic biomarkers in patient serum and found significant declines in soluble VEGF receptor, leptin, and PDGF-alpha, which suggested anti-tumor activity, and a significant increase was observed in an anti-angiogenic marker, placental growth factor (P1GF). In addition, analysis of serum biochemical markers of bone formation and resorption showed that bone-specific alkaline phosphatase increased and N-telopeptide decreased after treatment, suggesting that treatment with sunitinib may have an effect on reducing bone resorption. Due to the anti-angiogenic activity of sunitinib in CRPC, an international Phase III clinical trial of sunitinib has been launched in men with docetaxel-resistant metastatic prostate cancer (at www.clinicaltrials.gov - reference number NCT00676650).
Prostate-specific antigen (PSA) and radiological responses in prostate cancer patients 12 weeks after beginning sunitinib treatment. Each bar represents the change in serum PSA from baseline (day 1) to week 12 in individual patients. The bars are color coded based on radiographic response: partial response (PR-dark blue), stable disease (SD-red), progressive disease (PD-light blue), and not accessible (NE-yellow).
Publication:
Michaelson MD, Regan MM, Oh WK, et al. 2009. Phase II study of sunitinib in men with advanced prostate cancer. Annals of Oncology 20: 913-920.