
Lung Cancer



Posted March 30, 2018
Melin Khandekar, M.D., Ph.D., Massachusetts General Hospital

Dr. Melin Khandekar
One of the biggest challenges facing oncologists today is the development of treatment-resistant tumors. In many cases, a patient who starts off a treatment regimen with a positive response and tumor reduction experiences significant setbacks when the cancer develops resistance to the primary therapeutic. One approach to overcoming treatment resistance is to use a combination of therapies at the start of treatment to increase the sensitivity of the tumor to common therapeutics. This allows a chance for the original therapy to work before the tumor is able to adapt and develop resistance. Dr. Melin Khandekar, a Lung Cancer Research Program (LCRP) researcher at Massachusetts General Hospital, is using this approach to improve tumor response to commonly used chemotherapies.
Previous work at Dr. Khandekar's institution revealed that tumor cells tend to overexpress the peroxisome proliferator activator receptor-? (PPAR?), a receptor protein that plays a key role in metabolism and is a popular target for antidiabetic drugs. Because of this overexpression, tumor cells were sensitized to treatment by targeting PPAR? using thiazolidinedione (TZD) class drugs, a common antidiabetic. Unfortunately TZDs have fallen out of use due to concerns about possible side effects. Through a Fiscal Year 2014 Career Development Award from the LCRP, Dr. Khandekar's work is focusing on testing the effectiveness of TZD alternatives and confirming that targeting PPAR? with these new compounds sensitizes cancer cells to carboplatin chemotherapy.
In a paper recently published in the Proceedings of the National Academy of Sciences, Dr. Khandekar shares his most recent findings. He first confirmed that one contributor to chemotherapy/DNA damaging agent resistance is the phosphorylation of an amino acid on PPAR?. Upon exposure to carboplatin, cancer cells respond by phosphorylating a serine residue (S273) of PPAR?. This decreases the cells' accumulation of DNA damage when exposed to chemotherapy, preventing cell death. Dr. Khandekar and his team confirmed that blocking the phosphorylation of S273 restores the accumulation of DNA damage from the chemotherapeutic agent, leading to apoptotic cell death. He approached this in two ways. The first was to mutate the serine that was being phosphorylated to an alanine (S273A). This mutation retains the activity of PPAR?, but prevents phosphorylation. The second approach used novel noncanonical agonist ligands (NALs), TZD alternatives, to inhibit the phosphorylation. Both techniques successfully resensitized the cancer cells to cytotoxic agents that directly target DNA.
Dr. Khandekar then investigated whether cells responding well to this combination treatment demonstrated a specific gene signature. He found a core set of 23 genes whose expression changes significantly when PPAR? phosphorylation is inhibited by mutation (S273A) and confirmed that most of these genes follow the same expression patterns in cells treated with carboplatin and NALs. Upon comparing this gene expression set to patient data, he found that patients with this particular gene signature tended to demonstrate better survival after chemotherapy than patients without it. One gene set within this signature that is particularly impacted by blocking PPAR? phosphorylation is the p53 pathway. The p53 pathway plays a key role in many cancers, arresting the cell cycle and facilitating DNA repair. Further investigation proved that phosphorylated PPAR? associates with p53, but non-phosphorylated PPAR? cannot interact with p53 with the same efficiency. Additionally, functional p53 is required for NALs to successfully resensitize cells to cytotoxic drugs, indicating that understanding the interaction of PPAR? and p53 is important to the advancement of these sensitizing therapies.
While a number of steps still need to be taken before these NALs are an option for patient treatment, this preclinical work shows significant promise. Because NALs are also appealing as potential antidiabetic drugs, they are more likely to be available to cancer patients sometime in the near future.
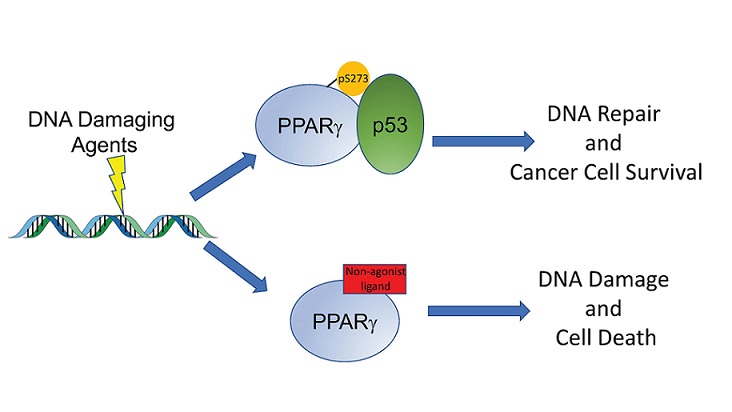
DNA Damaging agents such as chemotherapy cause phosphorylation of PPAR-gamma, which can then associate with the tumor suppressor P53, resulting in DNA repair and survival of cancer cells (upper arrow.) Treatment with a non-agonist PPAR-gamma ligand blocks this phosphorylation, which then disrupts the interaction with P53. The DNA damage is not repaired and accumulates, resulting in apoptotic cell death (lower arrow). Thus, these PPAR-gamma ligands can enhance the efficacy of many common anti-cancer therapies.
Reference:
Khandekar MJ, Banks AS, Laznik-Bogoslavski D, White JP, Choi JH, Kazak L, Lo JC, Cohen P, Wong KK, Kamenecka TM, Griffin PR, Spiegelman BM. 2018. PPAR? phosphorylation: A target for cancer therapy. Proceedings of the National Academy of Sciences, 115(3)561-566; DOI: 10.1073/pnas.1717776115.
Link:
Last updated Monday, March 10, 2025