 An official website of the United States government
An official website of the United States government
 ) or https:// means you've safely connected to the .mil website. Share sensitive information only on official, secure websites.
) or https:// means you've safely connected to the .mil website. Share sensitive information only on official, secure websites.
Duchenne Muscular Dystrophy
Toward a Safer and Better Gene Therapy for Duchenne Muscular Dystrophy
Posted September 6, 2023
Jeffrey Chamberlain, Ph.D., Julie Crudele, Ph.D., University of Washington

Dr. Julie Crudele
University of Washington

Dr. Jeffrey Chamberlain
University of Washington
Duchenne Muscular Dystrophy (DMD) is one of the most common genetic diseases, affecting around 1 in 3,500 to 5,000 male births worldwide. Since the discovery of the dystrophin gene in 1986,1 DMD has been an early candidate for gene therapy. It was not until June 22, 2023, when the U.S. Food and Drug Administration (FDA) announced accelerated approval of ELEVIDYS (delandistrogene moxeparvovec-rokl, an adeno-associated viral vector [AAV]-based gene therapy from Sarepta Therapeutics) for treatment of DMD, that the first gene therapy became available for patients affected with this devastating disease. It should be noted that the approval of ELEVIDYS was based on an increase in ELEVIDYS micro-dystrophin protein expression in skeletal muscle as a surrogate endpoint. Clinical functional evidence of ELEVIDYS treatment is still being validated through an ongoing phase 3 clinical trial. At the same time, acute serious liver injury, immune-mediated myositis and myocarditis have occurred in some patients treated with ELEVIDYS. Despite the still-to-be-affirmed efficacy and potential adverse events associated with this therapy, approval of ELEVIDYS, nonetheless, marks a milestone achievement for the concerted efforts from academia, industry, and advocacy groups over many decades. For Dr. Jeffrey Chamberlain, president of the American Society of Gene and Cell Therapy, whose expertise in viral vector design and genetic engineering contributed directly to the technology underlying the newly approved ELEVIDYS, this achievement marks only the first step toward better gene therapy strategies for DMD.
The dystrophin gene, one of the largest genes in the human genome (2.4 million base pairs), encodes the dystrophin protein, a transmembrane protein critical for the integrity and contractability of the skeletal muscles.2 Clinical variations of disease severity associated with different mutations or deletions of the dystrophin gene led to the initial understanding of how the different domains of the dystrophin protein contribute to its function. Further genetic engineering of shortened dystrophin fragments identified essential elements for dystrophin functionality, leading to the development of micro-dystrophin constructs containing only the essential coding regions of the dystrophin gene that can be effectively packaged into the AAV vectors. Based on significant efficacy in preclinical DMD animal models, five different micro-dystrophin constructs are currently in clinical trials for gene therapy on DMD, four of which are designed by Dr. Chamberlain's group,3 including the recently approved ELEVIDYS.
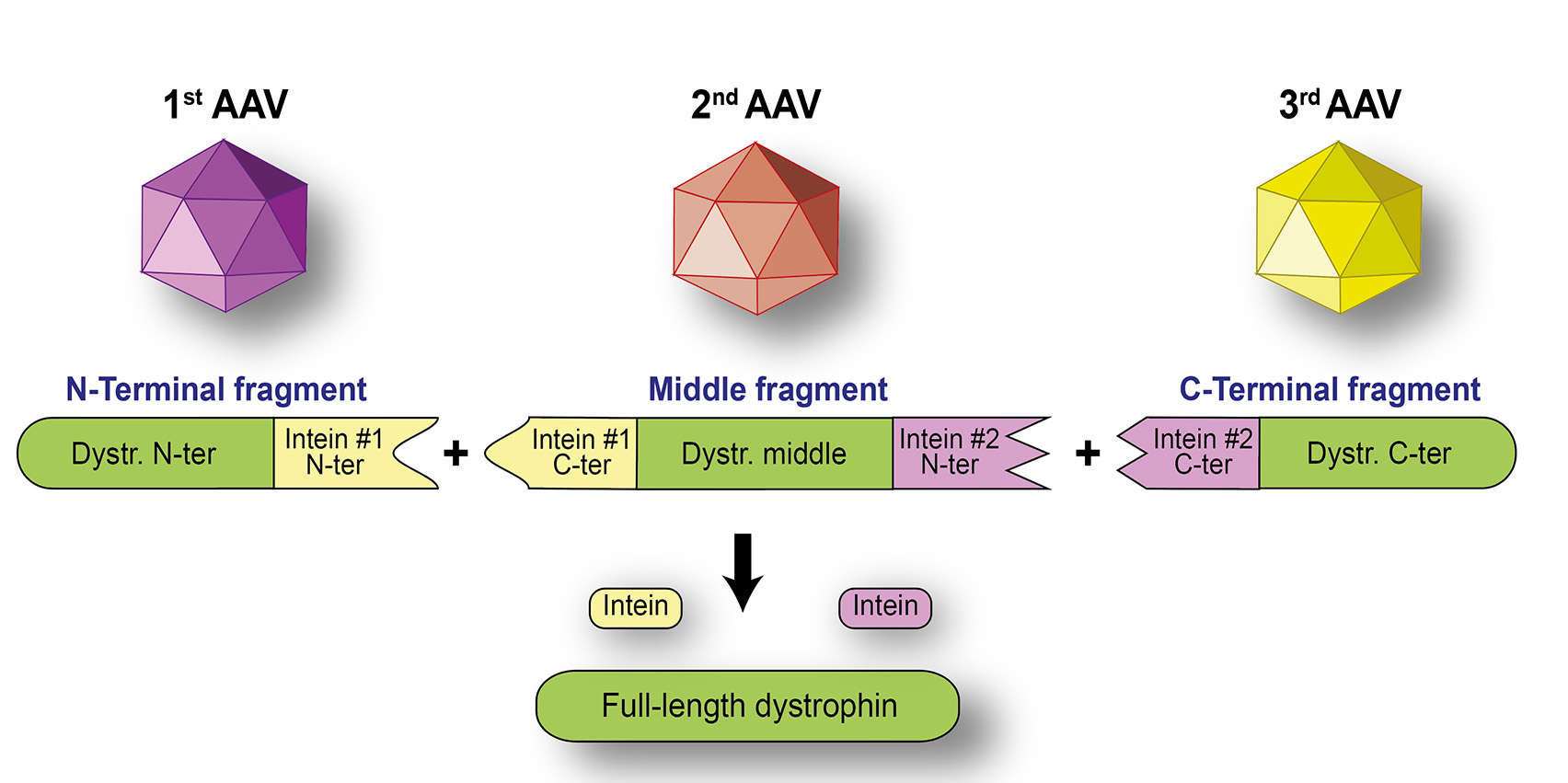 Figure 1. Full-length dystrophin can be joined by using two split-inteins
Figure 1. Full-length dystrophin can be joined by using two split-inteins
(Diagram provided by Dr. Jefferey Chamberlain)
While gene therapy strategies using micro-dystrophin are making strides toward clinical application for DMD, Dr. Chamberlain notes the clear limitations to such approaches. It is increasingly being recognized that efficacies from the truncated micro-dystrophin are variable, and clinical improvement is less than expected. The more closely the dystrophin constructs resemble the original gene, the better efficacy they can achieve as replacement gene therapy for DMD. In addition, severe adverse events arise due to immune reactivity to the large amount of AAV vector required to systemically target the muscles that make up 30%-40% of body mass. Using a Duchenne Muscular Dystrophy Research Program fiscal year 2022 Translational Research Award, in partnership with his mentee, Dr. Julie Crudele at the University of Washington-Seattle, Dr. Chamberlain will explore the possibility of using a lower dosage of split AAV vectors with much higher affinity for the muscles. These split AAV vectors will carry fragments of the dystrophin gene that will realign their protein products into larger pieces, or even full-length dystrophin in the muscle using the split-intein technique (Figure 1). Intein sequences are intervening protein sequences excised during transition from a protein precursor to a mature protein; split-inteins are capable of performing protein splicing when reconstituted into their active form.4 Computer-assisted protein structure redesign and deimmunization methods used in this project could allow seamless conjugation of two or more fragments of the protein products while minimizing potential immune-reactivity.
A natural yet significant step from the rational design of the micro-dystrophin constructs that gave rise to the first FDA-approved gene therapy, this new project led by
References:
1 Monaco AP, Neve RL, Colletti-Feener C, et al. 1986. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature Oct 16-22;323(6089):646-650. doi: 10.1038/323646a0. PMID: 3773991.
2 Gee SH, Madhavan R, Levinson SR, et al. 1998. Interaction of muscle and brain sodium channels with multiple members of the syntrophin family of dystrophin-associated proteins. Journal of Neuroscience Jan 1;18(1):128-137. doi: 10.1523/JNEUROSCI.18-01-00128.1998. PMID: 9412493; PMCID: PMC6793384.
3 Crudele JM and Chamberlain JS. 2019. AAV-based gene therapies for the muscular dystrophies. Human Molecular Genetics Oct 1;28(R1):R102-R107.
4 Shah NH and Muir TW. 2014. Inteins: Nature's gift to protein chemists. Chemical Science 5(1):446-461. doi: 10.1039/C3SC52951G. PMID: 24634716; PMCID: PMC3949740.
Last updated Tuesday, August 4, 2026