
Bone Marrow Failure



Posted May 22, 2018
Omar Abdel-Wahab, M.D., Memorial Sloan Kettering Cancer Center
Robert Bradley, Ph.D., Fred Hutchinson Cancer Research Center

Dr. Omar Abdel-Wahab

Dr. Robert Bradley
Myelodysplastic syndrome (MDS), also referred to as a "bone marrow failure disease" is most common in adults over the age of 65 and occurs when blood-forming cells in the bone marrow develop abnormally. The abnormal blood cells are defective and are destroyed by the body, resulting in a low number of blood cells. About one-third of patients with MDS progress to a rapidly growing cancer called acute myeloid leukemia (AML). Treatments for MDS and AML involve bone marrow transplantation or intensive chemotherapy. However, these are often not an option due to patient age and poor health, so there is an urgent need for alternative treatment strategies.
Splicing factors are involved in splicing the RNA in order to allow for proper processing, and a mutation in one of these proteins can result in cancer-causing mis-splicing. Interestingly, mutations rarely occur in more than one copy of a single spliceosomal gene, which suggests that the wildtype gene is important for cell survival, and that cells can tolerate only a partial deviation from the normal splicing activity. Spliceosomal genes (genes encoding splicing factors) are the most common mutational targets in MDS. Drs. Abdel-Wahab and Bradley hypothesized that treatment of spliceosomal gene mutant cells with a spliceosome inhibitor drug may cause an overwhelming amount of splicing dysregulation, rendering them defective. With support from the Bone Marrow Failure Research Program through a fiscal year 2011 (FY11) Postdoctoral Fellowship Award (for Dr. Abdel-Wahab) and an FY15 Idea Development Award (shared by both Drs. Bradley and Abdel-Wahab), the two investigators sought to test this theory and develop therapeutic approaches to target the spliceosomal mutant MDS.
Dr. Abdel-Wahab's laboratory expressed a spliceosomal gene mutation in the Srsf2 gene, a mutation common in MDS and AML patients, in the bone marrow cells of a leukemia mouse model. The bone marrow cells were engineered to contain (1) one wildtype and one mutant Srsf2 gene copy or (2) only one mutant Srsf2 gene copy. The mice expressing both mutant and wildtype Srsf2 rapidly developed fatal bone marrow failure and eventually developed leukemia. Mice expressing only mutant Srsf2 displayed a decreased incidence of developing leukemia. This is likely due to the decreased survival of leukemia-promoting bone marrow cells expressing mutant Srsf2 in the absence of wildtype Srsf2 expression. This illustrates that wildtype Srsf2 expression is indeed important for the survival of cells with Srsf2 spliceosome mutations.
Next, the effects of a splicing inhibition drug, E7107, were explored in models that mimic patient Srsf2-mutant leukemia. Patient Srsf2 mutant leukemia expresses both mutant and wildtype copies of Srsf2. Treatment with E7107 resulted in a survival benefit and decreased leukemic burden in Srsf2-mutant leukemia, while any response to E7107 in leukemia wildtype for splicing was minimal. This was detected in both a mouse leukemia model and a patient-derived AML model.
Dr. Bradley's lab then investigated the mechanistic origins of the Srsf2-mutant-selective effects of E7107. E7107 treatment of mice caused widespread intron retention and exon skipping in leukemic cells in both wildtype and mutant SRSF2 expressing cells. However, the magnitude of splicing inhibition following E7107 treatment was greater in cells expressing mutated SRSF2, suggesting that the combination of Srsf2 mutation and E7107 treatment has larger implications on splicing in these cells - implications that are likely responsible for their cell death.
Drs. Bradely and Abdel-Wahab's findings are all the more important when taking into consideration the high frequency of Srsf2 mutations in MDS and AML, the adverse outcomes associated with Srsf2 mutations and the need for better therapeutics to target them. Their research has provided support for therapeutic implications for MDS and AML patients with genetic alterations in Srsf2. The use of a splicing inhibitor such as E7107 could provide alternative, less invasive, and less intensive treatment of malignancies with Srsf2 splicing mutations.
Excitingly, these results have contributed to a Phase 1 clinical trial of a novel RNA splicing inhibitor for MDS, sponsored by H3 Biomedicine, Inc. (NCT02841540). The study began in August 2016 and is scheduled for completion in March 2019. The objectives of the trial are to evaluate the safety and recommended dose of a splicing modulator, H3B-8800, for patients with MDS and AML. This study will provide the data needed to progress to a Phase 2 clinical trial that will test whether treatment with a splicing inhibitor provides therapeutic benefit for patients. With positive results, these trials would provide the evidence needed to support new Food and Drug Administration-approved drugs that could provide a much needed therapy to older MDS and AML patients in poor health who have few treatment options.
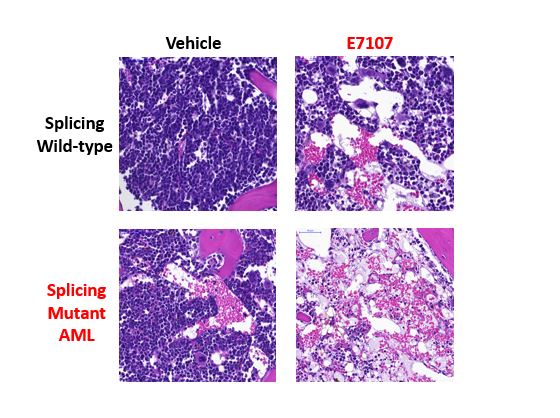
Bone sections from mice with AML in the SRSF2 wildtype (tops rows) or mutant (bottom rows) setting. In the left column are vehicle treated mice and in the right column are E7107-treated mice. The splicing inhibitor drug E7107 cleared leukemia cells most effectively and dramatically in the splicing mutant background.
Reference:
Lee SC, Dvinge H, Kim E, et al. 2016. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat Med. 22(6):672-678.
Links: